
5.1: Proteins
- Page ID
- 8395
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Proteins
Proteins are class of biomolecules that perform a wide array of functions in biological systems. Some proteins serve as catalysts for specific biochemical reactions. Other proteins act as signaling molecules that allow cells to "talk" with one another. Proteins, like the keratin in fingernails, can also act in a structural capacity. While the variety of possible functions for proteins is remarkably diverse, all of these functions are encoded by a linear assembly of amino acids, each connected to their neighbor via a peptide bond. The unique composition (types of amino acids and the number of each) and the order in which they are linked together determine the final three dimensional form that the protein will adopt and therefore, also the protein's biological "function". Many proteins can, in a cellular environment, spontaneously and often rapidly take on their final form in a process called protein folding. To watch a short (four minutes) introduction video on protein structure click here.
Protein structure
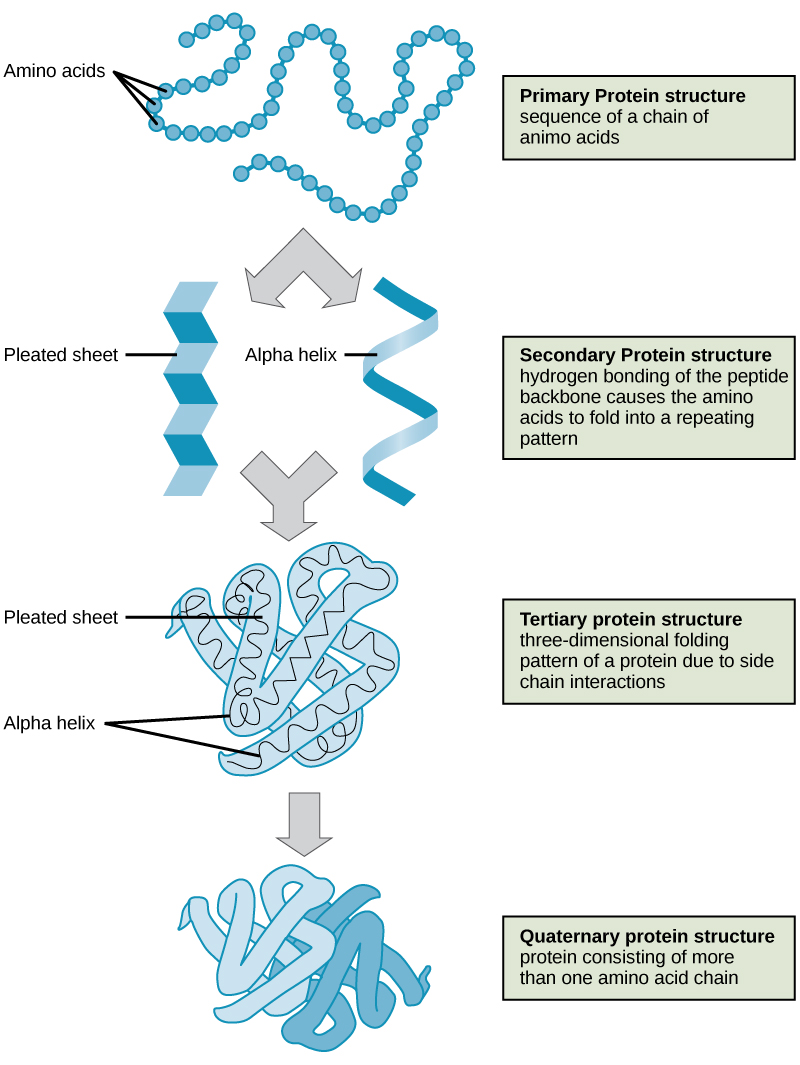
Protein structures can be described by four different levels of structural organization called primary, secondary, tertiary, and quaternary structures. These are briefly introduced in the sections that follow.
Primary structure
The unique sequence of amino acids in a polypeptide chain is its primary structure (Figure 1). The amino acids in this chain are linked to one another other via a series of peptide bonds. The chain of amino acids is often referred to as a polypeptide (multiple peptides).
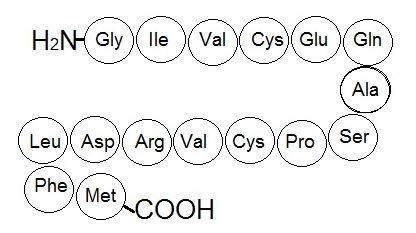
Due to the common backbone structure of amino acids, the resulting backbone of the protein has a repeating -N-Cα-C-N-Cα-C- pattern that can be readily identified in atomic resolution models of protein structures (Figure 2). Be aware that one of the learning goals for this class is for you to be able to examine a model like the one below and to identify the backbone from the side chain atoms (e.g. create the purple trace and blue shading if there aren't any). This can be done by finding the -N-Cα-C-N-Cα-C- pattern. Moreover, another learning goal for this class is that you are able to create drawings that model the structure of a typical protein backbone and its side chains (aka. variable group, R group). This task can be greatly simplified if you remember to start your model by first creating the -N-Cα-C-N-Cα-C- pattern and then filling in the variable groups.

Secondary structure
Due to the specific chemistry of the peptide bond the backbone between adjacent alpha-carbon atoms forms a highly planar structure (Figure 3). This means that all of the atoms linked by the pink quadrilateral lie on the same plane. The polypeptide is therefore structurally constrained since very little rotation can happen around the peptide bond itself. Rather, rotations occur around the two bonds extending away from the alpha carbons. These structural constraints lead to two commonly observed patterns of structure that are associated with the organization of the backbone itself.
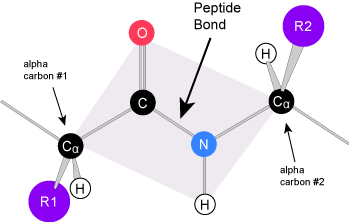
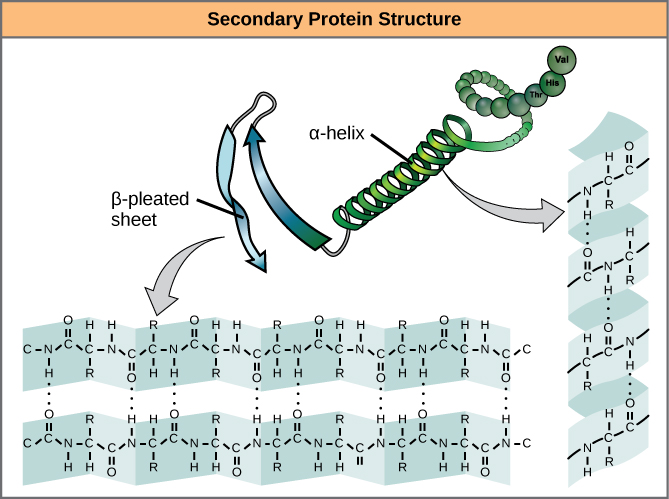
We call these patterns of backbone structure the secondary structure of the protein. The most common secondary structure patterns occurring via rotations of the bonds around each alpha-carbon, are the α-helix, β-sheet and loop structures. As the name suggests, the α-helix is characterized by a helical structure made by twisting the backbone. The β-sheet is actually the association between two or more structures called β-strands. If the orientation (N-terminus to C-terminus direction) of two associating β-strands are oriented in the same/parallel direction, the resulting β-sheet is called a parallel β-sheet. Meanwhile, if two associating β-strands are oriented in opposite/anti-parallel directions, the resulting β-sheet is called an anti-parallel β-sheet. The α-helix and β-sheet are both stabilized by hydrogen bonds that form between backbones atoms of amino acids in close proximity to one another. More specifically, the oxygen atom in the carbonyl group from one amino acid can form a hydrogen bond with a hydrogen atom bound to the nitrogen in the amino group of another amino acid. Loop structures by contrast refer to all secondary structure (e.g. backbone structure) that can not be identified as either α-helix or β-sheet.
Tertiary structure
The backbone and secondary structure elements will further fold into a unique and relatively stable three-dimensional structure called the tertiary structure of the protein. The tertiary structure is what we typically associate with the "functional" form of a protein. In Figure 6 two examples of tertiary structure are shown. In both structures, the protein is abstracted into a "cartoon" that depicts the polypeptide chain as a single continuous line or ribbon tracing the path between alpha carbons of amino acids linked to one another by peptide bonds - the ribbon traces the backbone of the protein (Figure 5).

The ribbon created by joining alpha-carbons can be drawn as a simple continuous line or it can be enhanced by uniquely representing secondary structural elements. For instance, when an α-helix is identified, the helix is usually highlighted by accentuating/broadening the ribbon to make the helical structure stand out. When a β-strand is present, the ribbon is usually broadened and an arrow is typically added to the C-terminal end of each β-strand - the arrow helps to identify the orientation of the polypeptide and whether β-sheets are parallel or anti-parallel. The thin ribbon connecting α-helix and β-strand elements is used to represent the loops. Loops in proteins can be highly structured and play an important role in the protein's function. They should not be treated lightly or dismissed as unimportant because their name lacks a Greek letter.

Attribution: Marc T. Facciotti (own work)
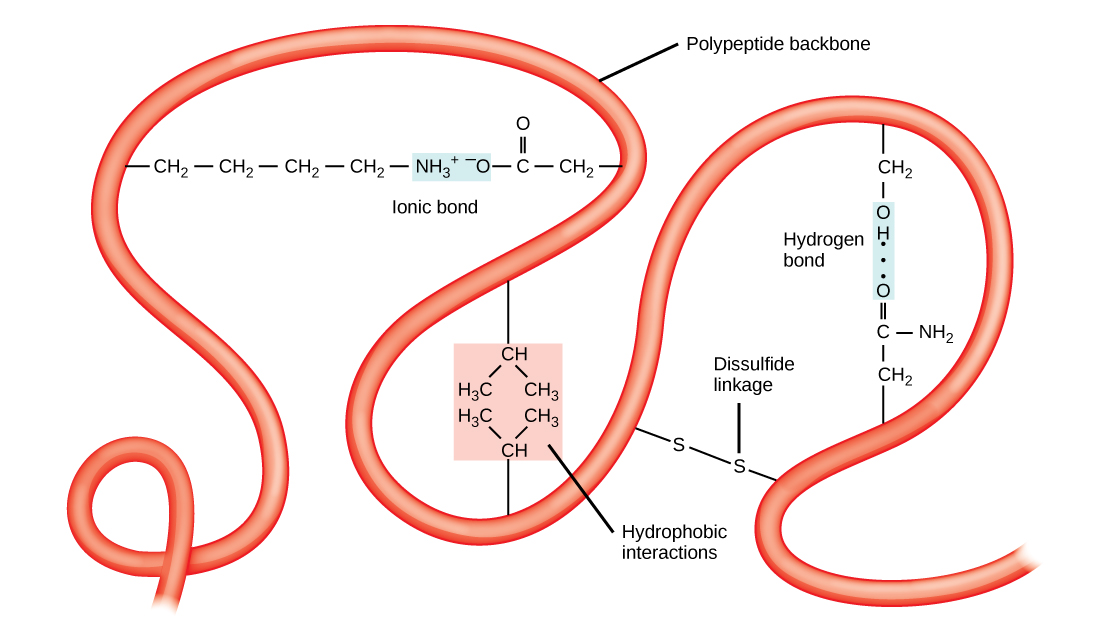
The tertiary structure is the product of many different types of chemical interactions among amino acid R groups, backbone atoms, ions in solution and water. These bonds include ionic, covalent, and hydrogen bonds and Van der Waals interactions. For example, ionic bonds may form between various ionizable side chains. It may, for instance, be energetically favorable for a negatively charged R group (e.g. an Aspartate) to interact with a positively charged R group (e.g. an Arginine). The resulting ionic interaction may then become part of the network of interactions that helps to stabilize the three dimensional fold of the protein. By contrast, R groups with like charges will likely be repelled by each other and be therefore unlikely to form a stable association thereby disfavoring a structure that would include that association. Likewise, hydrogen bonds may form between various R groups or between R groups and backbone atoms. These hydrogen bonds may also contribute to stabilizing the tertiary structure of the protein. In some cases covalent bonds may also form between amino acids. The most commonly observed covalent linkage between amino acids involves two cysteines and is termed a disulfide bond or disulfide linkage.
Finally, the association of the protein's functional groups with water also helps to drive chemical associations that help to stabilize the final protein structure. The interactions with water can, of course, include the formation of hydrogen bonds between polar functional groups on the protein and water molecules. Perhaps more importantly, however, is the drive for the protein to avoid placing too many hydrophobic functional groups in contact with water. The result of this desire to avoid interactions between water and hydrophobic functional groups means that the less polar side chains will often associate with one another away from water resulting in some energetically favorable Van der Waals interactions and the avoidance of energetic penalties associated with exposing the non-polar side chains to water. Indeed, the energetic penalty is so high for "exposing" the non-polar side chains to water that burying these groups away from water is thought to be one of the primary energetic drivers of protein folding and stabilizing forces holding the protein together in its tertiary structure.
Quaternary structure
In nature, the functional forms of some proteins are formed by the close association of several polypeptides. In such cases the individual polypeptides are also known as subunits. When the functional form of a protein requires the assembly of two or more subunits we call this level of structural organization the protein's quaternary structure. Yet again, combinations of ionic, hydrogen, and covalent bonds together with Van der Waals associations that occur through the "burial" of hydrophobic group at the interfaces between subunits help to stabilize the quaternary structures of proteins.
Denaturation
As was previously described, each protein has its own unique structure that is held together by various types of chemical interactions. If the protein is subject to changes in temperature, pH, or exposure to chemicals, that change the nature of or interfere with the associations between functional groups, the protein's secondary, tertiary and/or quaternary structures may change, even though the primary structure remains the same. This process is known as denaturation. While in the test tube denaturation is often reversible, in the cell the process can often be, for practical purposes, irreversible, leading to loss of function and the eventual recycling of the protein's amino acids. Resistance to environmental stresses that can lead to denaturation vary greatly amongst the proteins found in nature. For instance, some proteins are remarkably resistant to high temperatures; for instance, bacteria that survive in hot springs have proteins that function at temperatures close to the boiling point of water. Some proteins are able to withstand the very acidic, low pH, environment of the stomach. Meanwhile some proteins are very sensitive to organic solvents while others can be found that are remarkably tolerant of these chemicals (the latter are prized for use in various industrial processes).
Finally, while many proteins can form their three dimensional structures completely on their own, in many cases proteins often receive assistance in the folding process from protein helpers known as chaperones (or chaperonins) that associate with their protein targets during the folding process. The chaperones are thought to act by minimizing the aggregation of polypeptides into non-functional forms - a process that can occur through the formation of non-ideal chemical associations.