
2.5: DNA Replication
- Page ID
- 8422
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The scope of the problem
In this module we discuss the replication of DNA - one of the key requirements for a living system to reproduce or, in a multicellular system, to grow. Let us first briefly consider the scope of the problem by way of a literary analogy.
The human genome consists of roughly 6.5 billion base pairs of DNA if one considers the full diploid genome (i.e. if you count the DNA inherited from both parents). Six point five billion looks like this: 6,500,000,000. That's a large number. To get a better idea of what that number means, imagine that our DNA is a set of written instructions for constructing one of us. By analogy we can then compare it to another written document. For this example we begin by considering Tolstoy's "War and Peace", a novel many people are familiar with for its voluminous nature. Data from Wikipedia estimates that "War and Peace" contains about 560,000 words. A second written work many are familiar with are the seven volumes of J.K. Rowling's "Harry Potter". This work checks in at ~1,080,000 words (Referenced Statistics on Wikipedia). If we assume that the length of the average English word is 5 letters, the two literary works are 2.8 million and 5.4 million letters in length, respectively. Therefore, even all seven volumes of "Harry Potter" have over 1000x fewer letters than our own genomes. The number of letters in these 7 novels are, however, much closer to the number of nucleotides in a typical bacterial genome.
Now imagine yourself copying these texts. How fast could you do it? How many mistakes are you likely to make? Do you expect there to be a trade-off between the speed at which you can copy and the accuracy? What type of resources does this process need? How much energy is required? Now imagine copying something 1000x larger!
With that in mind, it is worth noting that a human cell can take about 24 hours to divide (DNA replication must therefore be a little faster). A healthy E. coli cell may take only 20 minutes to divide (including replicating its ~4.5 million base pair genome). Both the human and bacterium do this while typically making few enough mistakes that the subsequent generation remains viable and recognizable. That pretty amazing!
Design challenge
If the cell is to replicate - its ultimate goal - a copy of the DNA must be created. So one clear problem/statement/question is "how can the cell effectively copy its DNA?" Given the analogy above, some relevant sub-questions of relevance might be: What are the chemical and physical properties that enable DNA to be copied (we're not just building more DNA- we're building an exact copy of its sequence)? With what fidelity must the DNA be copied? What speed must it be copied at? Where does the energy come from for this task and how much is necessary? Where do the "raw materials" come from? How do the molecular machines involved in this process couple the assembly of raw materials and the energy required to build a new DNA molecule together? The list could, of course, go on.
In the following and in lecture we will discuss how the DNA replication is accomplished while keeping in mind some of these driving questions. As you go through the reading and lecture materials try to be constantly aware of these and other questions associated with this process. Use these questions as guideposts for organizing your thoughts and try to find matches between the "facts" that you think you might be expected to know and the driving questions.
The DNA Double Helix
To build some extra context we also need a little bit of empirically determined knowledge. Perhaps one of the best known and popular features of the hereditary form of the DNA molecule is that it has a double helical structure. The appreciation of this dates to the the 1950s. The story of this discovery has been widely recounted - and the details are beyond the scope of this text. Briefly, Francis Crick and James Watson are credited with determining the structure of DNA. Rosalind Franklin is now also widely credited with generating critical X-ray diffraction data that enabled Watson and Crick to piece together the puzzle of the DNA molecule.
Models of the structure of DNA revealed the molecule is made up of two strands of covalently linked nucleotides that are twisted around each other to form a right-handed helix. In each strand, nucleotides are covalently joined to two other nucleotides (except at the very ends of a linear strand) via phosphodiester bonds that link the sugars via the 5' and 3' hydroxyl groups (see panel b in the figure below) - recall that the labels 5' and 3' refer to the carbons on the sugar molecule. These sugar and phosphate chains form a contiguous set of covalent links that are often referred to as the "backbone" of the structure. In a linear molecule, each strand has two free ends. One is termed the 5'-end because the unlinked functional group that is typically involved in joining nucleotides is the phosphate linked to the 5' carbon. The other end of the strand is called the 3'-end because the unlinked functional group that is involved in joining nucleotides is the hydroxyl group linked to the 3' carbon of the sugar. Since the two ends of the strand are not symmetric this makes it easy to designate or describe a direction on the strand - one can, for instance, say that they are reading from the 5'-end to 3'-end to indicate that they are "walking" along the strand starting at the 5'-end and and moving towards the 3'-end. This direction (5' to 3') is, by the way, the convention used by biologists for writing sequences. The two strands of covalently linked nucleotides are found to be anti-parallel to one another in the double-helix; that is, the orientation/direction of one strand is opposite to that of the other strand (see panel b in the figure below). The sugar-phosphate backbone is on the outside of the double helix, creating a band of negative charges on the surface. By contrast, the nitrogenous bases of each of the antiparallel strands stack on the inside of the structure and oppose one another in a way that allows hydrogen bonds between unique purine/pyrimidine pairs (A pairing with T and G pairing with C) to form. "Stacking" refers to the fact that the flat planes of the bases on the same strand of DNA stack- like a stack of pancakes. These specific pairings (A with T, C with G) are said to be complementary to on another and thus the opposite strands of a double helix are often referred to one another as complementary strands.
Complementary strands carry redundant information. Because of the strict chemical pairing, if you know the sequence of one strand you also know the strand of its complement. Take for example the sequence 5′- C A T A T G G G A T G - 3′. Note how the sequence is annotated with the orientation (indicated by 5' and 3' labels). The complement of this sequence - written according to the 5' to 3' convention is: 5′- C A T C C C A T A T G - 3′. If you aren't convinced, write these two sequences out across from one another, making sure to write them as antiparallel strands. Note that the twisting of the two complementary strands around each other results in the formation of structural features called the major and minor grooves will become more important when we consider the binding of proteins to DNA (see panel c in the figure below).
Most of the Bis2a instructors will expect you to recognize key structural features depicted in the figure below and that you will be able to create a basic figure of the structure of DNA yourself.
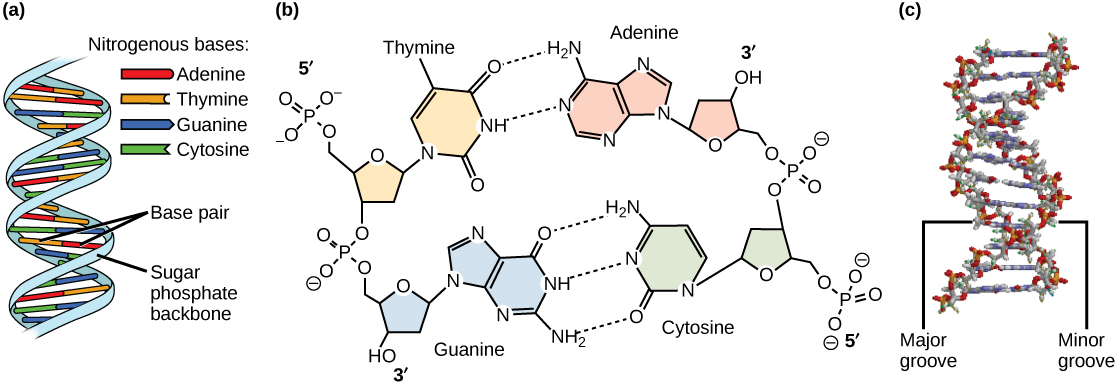
DNA has (a) a double helix structure and (b) phosphodiester bonds. The (c) major and minor grooves are binding sites for sequence-specific DNA binding proteins during processes such as transcription (the creation of RNA from a DNA template), the regulation of transcription, and the identification of origins of replication by specific proteins (note that DNA polymerase is not among these!). The winding of DNA around nucleosomes, in contrast, is not sequence-specific and simply involves electrostatic interactions between the negatively charged backbone and the positively charged outer surface of the nucleosome. Similarly, DNA polymerase will copy any sequence, and will bind to the 3' end of any legitimately base-paired primer.
The structure of DNA immediately suggested how DNA might replicate. The two strands could, in theory, separate, new nucleotides could be aligned in a specific order that is complementary to the template strand, and these new nucleotides could be joined together. Three competing models for replication were suggested: the conservative model, the semi-conservative model and the dispersive model.
- Conservative: The conservative model of replication postulated that each whole double-stranded molecule could act as a template for the synthesis of a completely new double-stranded molecule. That is, if one were to put a chemical tag on the template DNA molecule after replication none of that tag would be found on the new copy. This model seems a little silly, as the strands would have to separate, duplicate, and then separate again to find and re-bond to their original partner. Or a new strand would have to be formed side by side with an intact double helix, following some sort of templating that does not involve Watson-Crick base pairing.
- Semi-conservative: This hypothesis stipulated that each individual strand of a DNA molecule could serve as a template for a new strand, to which it would now be hydrogen bonded. In this case, if a chemical label were placed on the double stranded template DNA molecule, one strand on each of the copies would retain the label.
- Dispersive: This model proposed that a copied double-helix would be a piecewise combination of continuous segments of "old" and "new" strands. If a chemical label were placed on a DNA molecule that were copied using a dispersive mechanism, one would find discrete segments of the resulting copy that were labeled on both strands separated by completely unlabeled parts. This model, again, seems gratuitously complicated, requiring that the template strand be repeatedly broken and rejoined to newer molecules.
Meselson and Stahl resolved the issue in 1958 when they reported results of a now famous experiment which showed that DNA replication is semi-conservative (see figure below), and that each strand is used as a template for the creation of the new strand- as they no doubt expected. To learn more about this experiment watch The Meselson-Stahl Experiment.
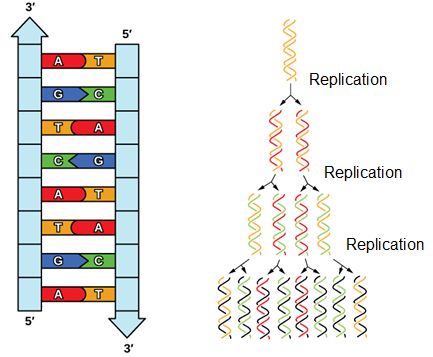
DNA has an anti-parallel double helix structure, the nucleotide bases are hydrogen bonded together and each strand complements the other. DNA is replicated in a semi-conservative manner, each strand is used as the template for the newly made strand.
DNA Replication
Having established some basic structural features and the need for a semi-conservative mechanism it is important to understand what is known about the process and to think about what questions one might want to answer.
Since DNA replication is a process we can invoke the "energy story" to think about it. Recall that an energy story is there to help us think systematically about processes (how things go from A to B). In this case the process is the act of starting with one double-stranded DNA molecule and ending up with two double-stranded DNA molecules. So, we will ask things like: What does the system look like at the beginning (matter and energy) of replication? How are matter and energy transferred in the system and what catalyzes the transfers? What does the system look like at the end of the process? We can also ask questions regarding specific events that MUST happen during the process. For instance, since DNA is a long molecule and it is sometimes circular, we can ask basic questions like, where does the process of replication start? Where does it end? We can also ask practical questions about the process like, what happens when a double-stranded structure is unwound?
We consider some of these key questions in this text and in class, and encourage you to do the same.
Requirements for DNA replication
Let's start by listing some basic functional requirements for DNA replication that we can infer just by thinking about the process that must happen and/or be required for the replication to happen. So, what do we need?
• We know that DNA is composed of nucleotides. If we are going to create a new strand we will need a source of nucleotides.
• We can infer that building a new strand of DNA will require an energy source - we should try to find this.
• We can infer that that there must be a process for finding a place to start replication.
• We can infer that there will be one or more enzymes that help catalyze the process of replication.
• We can also infer that, given the enormous number of bases to be copied, some mistakes will be made.
We will find that once we understand the basics of how replication occurs, additional questions/issues will be raised!
Nucleotide Structure
The building blocks of DNA are the nucleotides. Nucleotides are composed of a nitrogenous base, deoxyribose (a 5-carbon sugar), and a phosphate group. The nucleotide is named according to its nitrogenous base, purines such as adenine (A) and guanine (G), or pyrimidines such as cytosine (C) and thymine (T). Recall the structures below. Note that the nucleotide Adenosine triphosphate (ATP) is a precursor of the deoxyribonucleotide (dATP) which is incorporated into DNA. The other nucleotides to be employed during DNA synthesis are also nucleoside triphosphates. Hopefully this fact will provide some clues as to where the energy for DNA synthesis might come from.
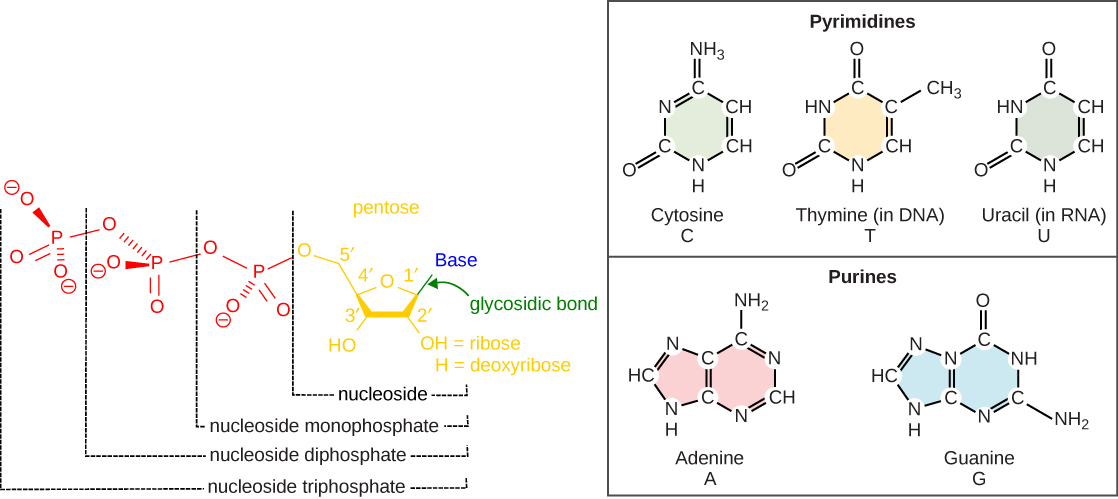
Each nucleotide is made up of a sugar (ribose or deoxyribose depending on whether it builds RNA or DNA, respectively), a phosphate group, and a nitrogenous base. The purines have a double ring structure with a six-membered ring fused to a five-membered ring. Pyrimidines are smaller in size; they have a single six-membered ring structure. The carbon atoms of the five-carbon sugar are numbered 1', 2', 3', 4', and 5' (1' is read as “one prime”). The phosphate residue is attached to the hydroxyl group of the 5' carbon of one sugar of one nucleotide and the hydroxyl group of the 3' carbon of the sugar of the next nucleotide, thereby forming a 5'-3' phosphodiester bond.
Initiation of Replication
With millions, if not billions, of nucleotides to copy, how does the DNA polymerase know where to start? Perhaps it might start anywhere, or, more reasonably, begin at one end of a chromosome and proceed to the opposite end? Neither of these hypotheses is correct. Evidence indicates that there are specific nucleotide sequences called origins of replication along the length of the DNA at which replication begins. The circular E. coli chromosome has just one of these sites; the linear eukaryotic chromosomes, in contrast, have multiple sites on every chromosome. Once this site is identified, however, there is a problem. The DNA double helix is held together by hydrogen bonds. If each strand is to be read and copied individually, there must be some mechanism responsible for dissociating the two strands. Breaking these hydrogen bonds is an endergonic process. Where does the energy come from and how is this reaction catalyzed? Basic reasoning should, at this point, lead to the hypothesis that a protein catalyst is involved and that this enzyme couples the endergonic separation of strands to some exergonic process.
It turns out that the details of this process and the proteins involved differ depending on the specific organism in question and many of the molecular level details are yet to be completely understood. There are, however, some common features in the establishment of replication origins in eukaryotes, bacteria and archaea. First, proteins generally called "initiators" have the capacity to bind DNA at or very near the DNA sequences that mark the origins of replication. The interaction of the initiator proteins with the DNA helps to destabilize the double helix and also to recruit other proteins, including an enzyme called a helicase. In this case the energy required to destabilize the DNA double helix seems to come from the formation of new associations between DNA and the initiator proteins. The DNA helicase, in contrast, once loaded onto the origin, couples the exergonic hydrolysis of ATP to the unwinding of the DNA double helix. Additional proteins must be recruited to the partially unwound initiation complex. These include, but are not limited to, enzymes called primase and DNA polymerase. While the initiators are lost soon after the initiation of replication, the rest of the proteins work in concert to execute the process of DNA replication. This complex of enzymes function at Y-shaped structures in the DNA called replication forks (see figure below). For any replication event two replication forks may be formed at each origin of replication, extending in both directions.
Suggested discussion
Why would different organisms have different numbers of replication origins? What could the benefit be to having more than one? Is there a drawback to having more than one?
Suggested discussion
Given what needs to happen at origins of replication, can you use logic to infer and propose for discussion some potential features that distinguish replication origins from other segments of DNA?
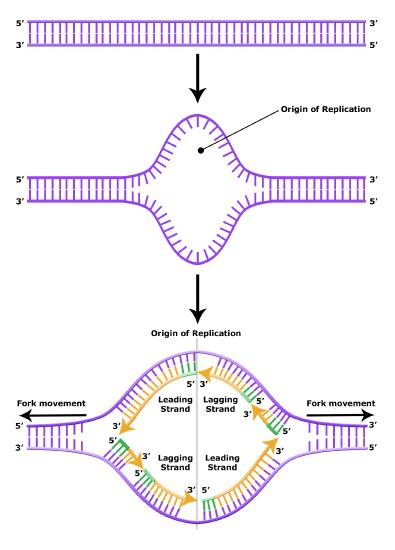
Elongation of Replication
The melting open of the DNA double helix and the assembly of the DNA replication complex is just the first step in the process of replication. Now the process of creating a new strand actually needs to get started. Here additional challenges are encountered. The first obvious issue is that of determining which of the two strands should be copied at any replication fork (i.e. which strand will serve as a template for semi-conservative synthesis)? Are both strands equally viable alternatives? There is also the problem of actually getting the process of new strand synthesis started. Can the DNA polymerase initiate a new strand on its own? It has been experimentally determined that DNA polymerase can NOT initiate strand synthesis. Rather, DNA polymerase requires a short stretch of double-stranded nucleic acids followed by single stranded template. DNA polymerase can only add a base to properly base-paired preexisting 3' end. This raises a bit of a problem for the initiation of DNA synthesis, doesn't it?
Fortunately, DNA polymerase can add a dNTP to an RNA molecule hybridized to a DNA template, and RNA polymerases do not require a preexisting base-paired 3' end tp initiate synthesis. Thus every molecule of DNA synthesis is actually initiated from a short (in E. coli, less than a dozen bases) RNA primer (these are depicted as short green lines in the figures above and below). The creation of a short primer is carried out by the enzyme primase. Primase is a very slow (well, relatively slow- requiring one second to make a primer) and error-prone polymerase. The error-prone nature of its activity is not a problem, because the cell will remove all the primers later in the process of DNA synthesis- and we'll revisit this later in this reading.
During the process of strand elongation, DNA polymerase polymerizes a new covalently-linked strand of DNA nucleotides (in E. coli this replicative polymerase is called DNA polymerase III; in eukaryotes polymerase nomenclature is more complex and the roles of the several replicative polymerases are not completely understood). DNA polymerase will ride along the template strand in that strand's 3' to 5' direction, synthesizing a new strand by adding bases to the nascent (new-born) strand's 3' end. (I suggest you make your own diagram to clarify this directionality- remember the two strands are antiparallel- the 3' to 5' direction on the template is the 5' to 3' direction on the new strand! If this sounds confusing, then get to work on that diagram asap).
Let us briefly consider the reaction involving the addition of a single nucleotide. The primer provides an important 3' hydroxyl on which to begin synthesis. The next deoxyribonucleotide triphosphate enters the binding site of the DNA polymerase and is oriented by the polymerase such that a hydrolysis of the incoming 5' triphosphate can occur, releasing pyrophosphate and coupling this exergonic reaction to the endergonic synthesis of a phosphodiester bond between the 5' phosphate of the incoming nucleotide and the 3' hydroxyl group of the primer. The degradation of pyrophosphate will add an extra energetic kick to this reaction. This process will then be repeated. There is no real "termination site" for DNA synthesis, synthesis by any individual polymerase will continue until the replication complex dissociates from the DNA; the complex might "fall off" the end of a broken template strand, it might run off the end of a linear chromosome, or it might run up against the 5' end of a previously synthesized primer (more on this later). This sounds much more complicated in text than it really is: see the "Leading and Lagging strand synthesis" diagram below (and definitely draw your own version).
Correct base pairing, or selection of correct nucleotide to add at each step, is accomplished by structural constraints felt by the DNA polymerase and the energetically favorable hydrogen bonds formed between complementary nucleotides. The process is energetically driven by the hydrolysis of the incoming 5' triphosphate and the energetically favorable interactions formed by the inter-nucleotide interactions in the growing double helix (base stacking and complementary base pairing hydrogen bonds).
After elongation of any particular new molecule is complete, a different DNA polymerase (in bacteria this is usually called DNA Polymerase I) comes in to remove the RNA primer and to synthesize the remaining bit of missing DNA. It will use the 3' end recently of the new strand recently created by DNA polymerase III as its primer.
The movement of the replication fork, and the separation of strands by DNA helicase, induces over-winding of the DNA in both ahead of the fork (imagine taking two strings twisted around each other, and trying to peel them apart- you'd end up with a tangle in the unseparated portion). Another ATP consuming enzyme called, in E. coli, gyrase, helps to relieve this stress. This enzyme is located just ahead of the replication fork, and repeatedly nicks and rejoins the DNA, allowing the supercoils to relax.
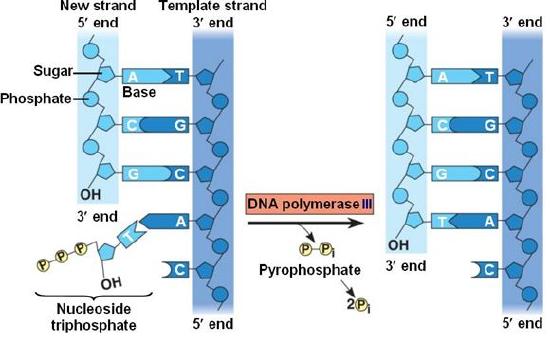
DNA polymerase catalyzes the addition of the 5' phosphate group from an incoming nucleotide to the 3' hydroxyl group of the previous nucleotide. This process creates a phosphodiester bond between the nucleotides while hydrolyzing the phosphoanhydride bond in the nucleotide.
Source: http://bio1151.nicerweb.com/Locked/m...h16/elong.html
Suggested discussion
Create an energy story for the addition of a nucleotide onto a polymer as shown in the figure above. This will be an explicit learning goal from some of your Bis2A instructors.
Leading and Lagging Strand
The discussion above about strand elongation describes the process of new strand synthesis if that strand happens to be synthesized in the same direction as the replication fork is or appears to be moving along the DNA. This strand can be synthesized continuously and is called the leading strand (and it moves along the "leading strand template"). However, both strands of the original DNA double helix must be copied, and to minimize the accumulation of single-stranded DNA (which is both susceptible to damage and difficult to repair), both strands are replicated simultaneously. Since the DNA polymerase can only synthesize DNA in a 5' to 3' direction, the polymerization of the strand opposite of the leading strand must occur in the opposite direction that the replication fork is traveling (this would be a good time to try to draw all of this, to orient yourself). This strand is called the lagging strand and due to geometric constraints must be synthesized through a repeated series of RNA priming and DNA synthesis events, creating short segments of new DNA called Okazaki fragments. As noted, the initiation of synthesis of each Okazaki fragment requires primase to synthesize an RNA primer and each of these RNA primers must be ultimately removed and replaced with DNA nucleotides by a different DNA polymerase. DNA polymerase I performs this job; it binds to the 3' end of an existing Okazaki fragement, using this (DNA) 3' to initiate replication. DNA polymerase possesses a 5' to 3' exonuclease activity and destroys the RNA nucleotides (former primers) in front of it, replacing them with dNTPs. This both eliminates RNA from the new strands. This is good for many reaons: as RNA is relatively unstable, primase is very error prone, and DNA polymerase would not be able to use RNA as a template for the next round of replication). The covalent bonds between each of the Okazaki fragments must therefore be formed by yet another enzyme called DNA ligase, which uses up an ATP to ligate the 3' end of the fragment to the 5' end of the (now RNA-free) Okazaki fragment in front of it.
Quick question- why is an ATP needed for DNA ligase to join two Okazaki fragments together? ATP's aren't needed for DNA polymerase to add a nucleotide...
The geometry of lagging strand synthesis is difficult to visualize and will be covered in class. However, this might be best represented by a video (one that moves slowly!). A realistic representation of simultaneous leading and lagging strand synthesis can be found among Drew Barry's videos, here. However, this animation moves quickly (at actual speed). A really fascinating aspect of replication fork progression is the fact that the two polymerases, on the leading and lagging strand, are tethered together. They are both moving in the same direction as the replication fork, and the lagging strand template has to repeatedly twist and coil to accommodate this directionality. You will also see in this video how DNA polymerase has to be repeatedly reloaded and then dissociated from the lagging strand template. Both polymerases are stabilized on their template strands by a clamp that encircles that strand. In the video, you will see the "clamp loader" as a large complex that repeatedly grabs and reloads green clamps from the nucleoplasm.
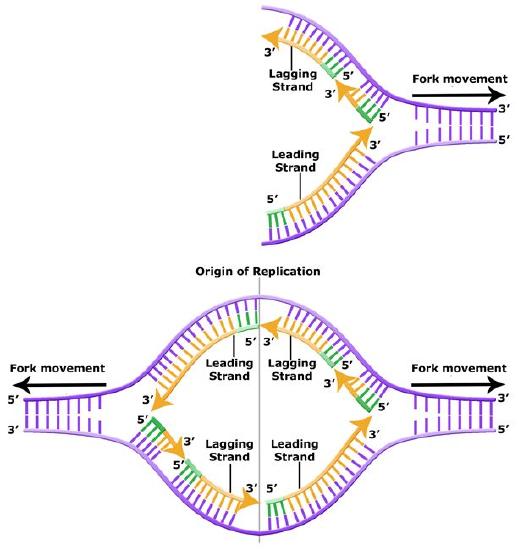
Leading and lagging strand synthesis. The lagging strand is created in multiple segments. A replication fork showing the leading and lagging strand. A replication bubble showing the leading and lagging strands.
Bis2A Team original image
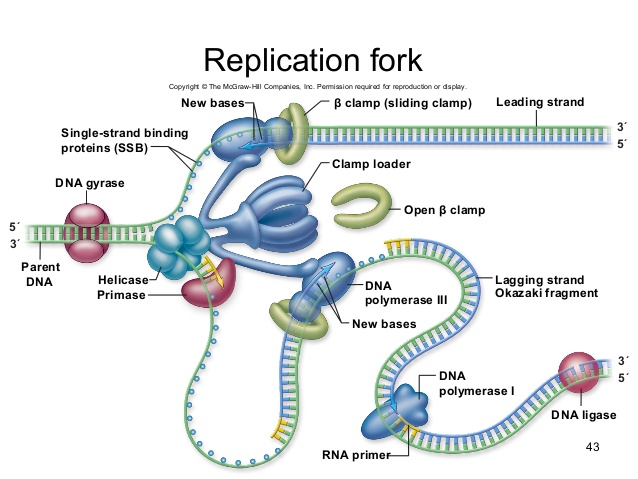
A growing replication fork. All the enzymes required for DNA replication (in E. coli)- with the exception of initiation- are illustrated here, although in a very stylized (but very clear) fashion. Which components are involved in leading and lagging strand priming? Strand extension? Removal of RNA nucleotides (former primers). Joining of separate Okazaki fragments? Coping with accumulation of supercoils? Note that the leading and lagging strand polymerases are both tethered to the clamp loader. How can DNA be simultaneous synthesized on both the leading and lagging stands if the polymerases are bound to each other?
Termination of Replication
An issue specific to linear chromosomes: Telomeres and Telomerase
The termination of replication in circular bacterial chromosomes poses few practical problems. However, the ends of linear eukaryotic chromosomes pose a specific problem for DNA replication. Because DNA polymerase can add nucleotides in only one direction (5' to 3'), the leading strand allows for continuous synthesis until the end of the chromosome is reached; however, as the replication complex arrives at the end of the lagging strand there is no place for the primase to "land" and synthesize an RNA primer so that the synthesis of the missing lagging strand DNA fragment at the end of the chromosome can be initiated by the DNA polymerase. Without some mechanism to help fill this gap, this chromosomal end will remain unpaired and the the chromosome will become progressively shorter with each round of replication, ultimately compromising the ability of the organism to survive. These ends of the linear chromosomes are known as telomeres and contain highly repetitive, very short sequences that do not code for proteins. As a consequence, these "non-coding" telomeres act as replication buffers and, in somatic cells, are indeed shortened with each round of DNA replication. For example, in humans, a six base-pair sequence, TTAGGG, is repeated 100 to 1000 times at the end of most chromosomes. Clearly this is a stop-gap solution! The discovery of the enzyme telomerase helped in the understanding of how chromosome ends are maintained. Telomerase is an enzyme composed of protein ( a reverse transcriptase, meaning, an enzyme that copies an RNA template to make DNA) and a short RNA template. Telomerase attaches to the end of the chromosome by complementary base pairing between the RNA component of telomerase and the telomeric sequence of the DNA. The RNA is then used as template for the elongation of the chromosome end. This process can be repeated numerous times. Once the lagging strand template is sufficiently elongated by telomerase, primase will create a primer followed by DNA polymerase which can now add nucleotides that are complementary to the ends of the chromosomes. Thus, the ends of the chromosomes are replicated.
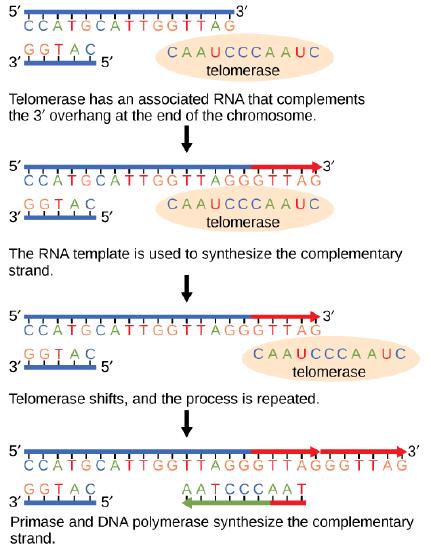
The ends of linear chromosomes are maintained by the action of the telomerase enzyme.
Telomerase is not active in somatic cells. Adult somatic cells that undergo cell division continue to have their telomeres shortened. Telomerase is, however, expressed in germline cells- cells that will ultimately produce gametes. Thus each zygote inherits chromosomes with long telomeres. Mutants completely defective in telomerase function often have no phenotype, but as the generations progress, their offspring become increasingly defective. This is due to the gradual loss of telomeric sequences with each cell division; once these sequences are lost the chromosomes become extremely unstable and exhibit frequent breakage. Thus telomeric sequences not only keep chromosomes long, they also prevent chromosome ends from being mistakenly recognized as "chromosome breaks". The instability of telomere-less chromosomes is due to the cell's misguided attempts to "repair" these breaks.
Differences in DNA Replication Rates Between Bacteria and Eukaryotes
DNA replication has been extremely well-studied in bacteria, primarily because of the small size of the genome and large number of variants available. Escherichia coli has 4.6 million base pairs in a single circular chromosome, and all of it gets replicated in approximately 42 minutes, starting from a single origin of replication and proceeding around the chromosome in both directions. This means that approximately 1000 nucleotides are added per second. The process is much more rapid than in eukaryotes.
| Property | Prokaryotes | Eukaryotes |
|---|---|---|
| Origin of replication | Single | Multiple |
| Rate of polymerization per polymerase | 500 nucleotides/s | 50 to 100 nucleotides/s |
| Chromosome structure | circular | linear |
| Telomerase | Not present | Present |
Link to external resource
Click through a tutorial on DNA replication.
Replication Design Challenge: Proofreading
When the cell begins the task of replicating the DNA, it does so in response to environmental signals that tell the cell it is time to divide. The goal of DNA replication is to produce two identical copies of the double-stranded DNA template and to do it in an amount of time that does not pose an unduly high evolutionarily selective cost. This is a daunting task when you consider that there are ~6,500,000,000 base pairs in the human genome and ~4,500,000 base pairs in the genome of a typical E. coli strain and that Nature has determined that the cells must make copies of themselves within 24 hours and 20 minutes, respectively. In either case many individual biochemical reactions need to take place.
While ideally replication would happen with perfect fidelity, DNA replication, like all other biochemical processes, is imperfect - bases may be left out, extra bases added, or bases may be added that do not properly base-pair. In fact, the difference in potential energy between some correctly base paired and incorrectly base paired nucleotides is simply insufficient to "power" the remarkable fidelity of DNA polymerase (which erroneously inserts nucleotides at a rate of less than 1/106). Many of the mistakes that occur during DNA replication are promptly corrected by DNA polymerase itself via a mechanism known as proofreading. In proofreading, the DNA polymerase "reads" each newly added base via sensing the presence or absence of small structural anomalies before adding the next base to the growing strand. In so doing, a correction can be made.
If the polymerase detects that a newly added base has paired correctly with the base in the template strand, the next nucleotide is added. If, however, an incorrect nucleotide is added to the growing polymer, the mis-shaped double helix will cause DNA polymerase to stall, and the newly made strand will be ejected from the polymerizing site on the polymerase and move into an exonuclease site. In this site, DNA polymerase is able to cleave off the last several nucleotides that were added to the polymer. Once a few nucleotides have been removed, new ones will be added again. This proofreading capability comes with some trade-offs: Using an error correcting/more accurate polymerase requires time. The slower you go the more accurate you can be. Going too slow, however, may keep you from replicating as fast as your competition, so figuring out the balance is key.
Errors that are not corrected by proofreading or mismatch repair (see below) may become mutations- but only if their presence is not detected by yet another quality-control process: mismatch repair (discussed below).
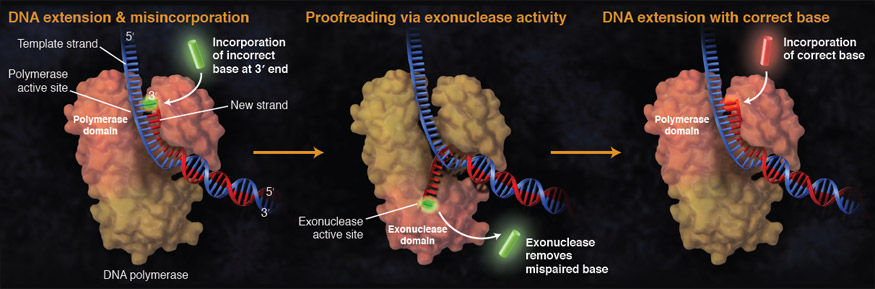
Suggested discussion
What are the pros and cons for DNA polymerases' proofreading capabilities?
Leading and lagging strand synthesis is complicated! Why not have a second DNA polymerase that extends the strand in the 3' to 5' direction instead? Having two polymerases, yoked together and proceeding on both strands simultaneously would seem to be a simpler solution. The energy requirement would still be fulfilled: to run polymerization in the opposite direction we'd simply have a growing strand that ends with a 5' triphosphate, which would be attacked by the 3' OH of the incoming dNTP. However, the imaginary polymerase that adds to the 5' end, rather than 3' end of the chain would have issues with proofreading. Their most recently added nucleotide carries the triphosphate. If this base is erroneous, and excised by the proofreading exonuclease, there will no longer be a triphosphate at the 5' end of the chain. Therefore, the chain could not be elongated. Try drawing this situation, for a real polymerase vs. this imaginary polymerase that elongates the 5' end of the growing chain.

Replication Mistakes and DNA Repair
Although DNA replication is typically a highly accurate process and proofreading DNA polymerases help to keep the error rate low (down to about 1/106 bases), mistakes still occur. In addition to errors of replication, environmental damage may also occur to the DNA. Such uncorrected errors of replication or environmental DNA damage may lead to serious consequences. Therefore, Nature has evolved several mechanisms for detecting and repairing damaged or incorrectly synthesized DNA.
Mismatch Repair
Some errors are not corrected during replication, but are instead corrected after replication is completed; this type of repair is known as mismatch repair. Specific enzymes recognize the incorrectly added nucleotide and excise it; replacing it with the correct base, using the sister strand as a template. Simple enough! The issue is: how do mismatch repair enzymes recognize which of the two improperly paired bases is the incorrect one?
In E. coli, at some point after replication, a subset of adenines (at a specific 4 base sequence) acquires a methyl group. Immediately after replication the parental (old) DNA strand will have methyl groups on these A's, whereas the newly synthesized strand lacks them (the enzyme that recognizes these sites hasn't found the new strand yet). Thus, this is a window of opportunity for mismatch repair enzymes are able to scan the DNA remove the wrongly incorporated bases from the newly synthesized, the non-methylated strand, using the methylated strand as the "correct" template from which to incorporate a new nucleotide. In eukaryotes, the mechanism for distinguishing old vs. new strands is not as well understood, but it is believed to involve recognition of unsealed nicks in the new strand, as well as a short-term continuing association of some of the replication proteins with the new daughter strand after replication has completed.
Nucleotide Excision Repair
Nucleotide excision repair enzymes replace damaged bases by making a cut on both the 3' and 5' ends of the damaged site. The entire segment of DNA is removed and replaced with correctly paired nucleotides by the action of a DNA polymerase. Once the bases are filled in, the remaining gap is sealed with a phosphodiester linkage catalyzed by the enzyme DNA ligase. This repair mechanism is often employed when UV exposure causes the formation of pyrimidine dimers.
Nucleotide excision repairs thymine dimers. When exposed to UV, pyrimidines lying adjacent to each other can form dimers- this distorts the helix and is not an acceptable template for DNA synthesis by the replicative polymerase. In normal cells, they are excised and replaced.
Reversal of damage
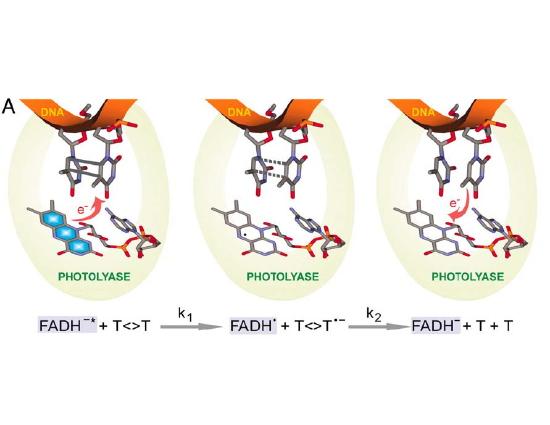
The mechanisms described above reflect a "remove and replace" strategy for repair of DNA, and take advantage of the redundancy in information of the double helix. Note that excision repair can't be employed in a single-stranded DNA or RNA genomes, which are frequently found in viruses. There are, however, a couple of types of damage that are so common that a specific repair pathway has evolved to recognize and simply reverse this specific type of damage to bases, rather than excising and replacing a damaged oligonucleotide. One example is photolyase- an enzyme that recognizes and reverses pyrimidine dimers, the most common form of UV induced damage. This enzyme is actually "powered" by blue light (which should be present in any environment where you would find UV light). The protein recognizes pyrimidine dimers and, when struck by light, donates (and then retrieves) an electron, breaking the inappropriate bonds between the two adjacent bases. Almost all living things express this enzyme- except, unfortunately, placental mammals. We're "stuck" with excision repair as our only mode of repair for UV-induced damage.
Consequences of errors in replication, transcription and translation
Something to think about:
Cells have evolved a variety of ways to make sure DNA errors are both detected and corrected. We have already discussed several of them. Why did these evolve? Such mechanisms did not evolve to repair RNA or proteins. What the consequences would be of an error in transcription? Would such an error effect the offspring? Would it be lethal to the cell? What about errors in translation? How do these contrast with errors in DNA replication? If you are not familiar with transcription or translation, don't fret. We'll learn those soon and return to this question again.