
Protein Composition and Structure
- Page ID
- 1346
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Secondary Structure of a Polypeptide Chain
Secondary structure refers to the shape of a folding protein due exclusively to hydrogen bonding between its backbone amide and carbonyl groups. Secondary structure does not include bonding between the R-groups of amino acids, hydrophobic interactions, or other interactions associated with tertiary structure.
The two most commonly encountered secondary structures of a polypeptide chain are alpha-helices and beta-pleated sheets. These structures are the first major steps in the folding of a polypeptide chain, and they establish important topological motifs that dictate subsequent tertiary structure and the ultimate function of the protein.
Ramachandran plot

http://commons.wikimedia.org/wiki/Fi...neral_100K.jpg
α-Helices
An alpha-helix is a right-handed coil of amino-acid residues on a polypeptide chain, typically ranging between 4 and 40 residues. This coil is held together by hydrogen bonds between the oxygen of C=O on top coil and the hydrogen of N-H on the bottom coil. Such a hydrogen bond is formed exactly every 4 amino acid residues, and every complete turn of the helix is only 3.6 amino acid residues. This regular pattern gives the alpha-helix very definite features with regards to the thickness of the coil and the length of each complete turn along the helix axis.
The structural integrity of an alpha-helix is in part dependent on correct steric configuration. Amino acids whose R-groups are too large (tryptophan, tyrosine) or too small (glycine) destabilize alpha-helices. Proline also destabilizes alpha-helices because of its irregular geometry; its R-group bonds back to the nitrogen of the amide group, which causes steric hindrance. In addition, the lack of a hydrogen on Proline's nitrogen prevents it from participating in hydrogen bonding.
Another factor affecting alpha-helix stability is the total dipole moment of the entire helix due to individual dipoles of the C=O groups involved in hydrogen bonding. Stable alpha-helices typically end with a charged amino acid to neutralize the dipole moment.
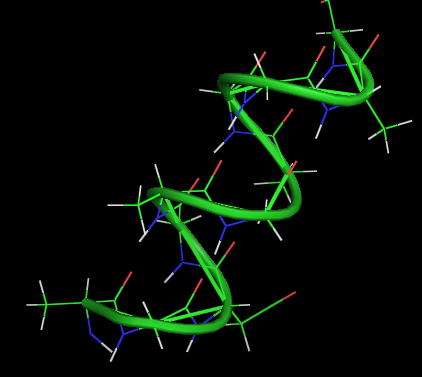
α-helix
- 3.6 amino acids per turn
- 0.54 nm per turn
- side chains pointed out
- H-bonds parallel to axis
- n-4 H-bonds
- dipole moment (neg. at C end)
- no pro, less gly, ser
- limited similar side chain charges
β-PLEATED SHEETS
This structure occurs when two (or more, e.g. psi-loop) segments of a polypeptide chain overlap one another and form a row of hydrogen bonds with each other. This can happen in a parallel arrangement:

Or in anti-parallel arrangement:
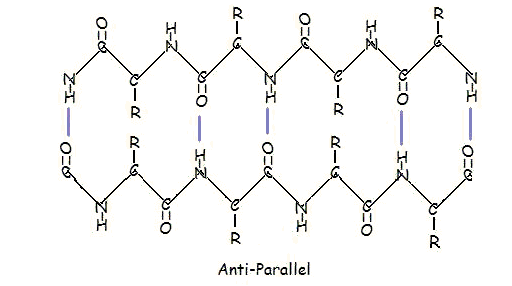
Parallel and anti-parallel arrangement is the direct consequence of the directionality of the polypeptide chain. In anti-parallel arrangement, the C-terminus end of one segment is on the same side as the N-terminus end of the other segment. In parallel arrangement, the C-terminus end and the N-terminus end are on the same sides for both segments. The "pleat" occurs because of the alternating planes of the peptide bonds between amino acids; the aligned amino and carbonyl group of each opposite segment alternate their orientation from facing towards each other to facing opposite directions.
The parallel arrangement is less stable because the geometry of the individual amino acid molecules forces the hydrogen bonds to occur at an angle, making them longer and thus weaker. Contrarily, in the anti-parallel arrangement the hydrogen bonds are aligned directly opposite each other, making for stronger and more stable bonds.
Commonly, an anti-parallel beta-pleated sheet forms when a polypeptide chain sharply reverses direction. This can occur in the presence of two consecutive proline residues, which create an angled kink in the polypeptide chain and bend it back upon itself. This is not necessary for distant segments of a polypeptide chain to form beta-pleated sheets, but for proximal segments it is a definite requirement. For short distances, the two segments of a beta-pleated sheet are separated by 4+2n amino acid residues, with 4 being the minimum number of residues.
- R2 (C=O side) is often G,A
- R3 (N-H side) is often D
- Proline is often R2 or R3
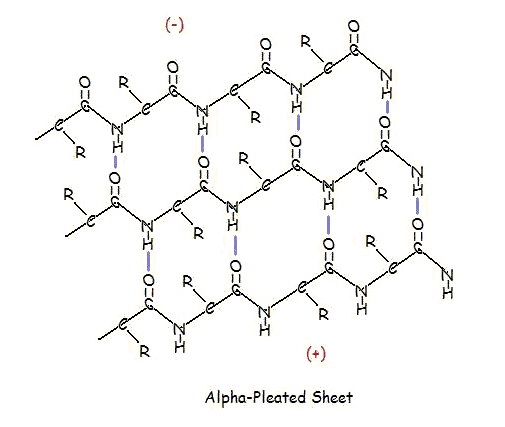
α-PLEATED SHEETS
A similar structure to the beta-pleated sheet is the alpha-pleated sheet. This structure is energetically less favorable than the beta-pleated sheet, and is fairly uncommon in proteins. An alpha-pleated sheet is characterized by the alignment of its carbonyl and amino groups; the carbonyl groups are all aligned in one direction, while all the N-H groups are aligned in the opposite direction. The polarization of the amino and carbonyl groups results in a net dipole moment on the alpha-pleated sheet. The carbonyl side acquires a net negative charge, and the amino side acquires a net positive charge.
Tertiary Structure

http://commons.wikimedia.org/wiki/Fi..._Structure.png
Quaternary Structure
The folding of a protein reduces the free energy (ΔG) of the system.
The folding of a protein involves both protein and solvent.