
10: Green Fluorescence Protein (GFP) Purification
- Page ID
- 169774
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Green Fluorescence Protein (GFP) Purification
– Hydrophobic Interaction Chromatography –
For the past years, the green fluorescent protein (GFP) from the jellyfish Aequorea victoria has become one of the most widely studied and exploited proteins in biochemistry and cell biology. Its amazing ability to generate a highly visible, efficiently emitting internal fluorophore is both intrinsically fascinating and tremendously valuable. High-resolution crystal structures of GFP offer unprecedented opportunities to understand and manipulate the relation between protein structure and spectroscopic function. GFP has become well established as a marker of gene expression and protein targeting in intact cells and organisms. Mutagenesis and engineering of GFP into chimeric proteins are opening new vistas in physiological indicators, biosensors, and photochemical memories.
Solutions:
TE buffer 10 mM Tris, 1 mM EDTA (pH 8.0)
Equilibration buffer TE plus 2.0 M (NH4)2SO4 (pH 8.0)
Binding buffer TE plus 4.0 M (NH4)2SO4 (pH 8.0)
Wash buffer TE plus 1.3 M (NH4)2SO4 (pH 8.0)
Growing Cell Cultures (Pre-lab preparation)
1. Remove the transformation plates from the incubator and examine using the UV light. Identify several green colonies that are not touching other colonies on the LB/amp plate. Identify several white colonies on the LB/amp plate.
2. Obtain two culture tubes containing the growth media LB/amp/ara. Label one "+" and one "-". Using a sterile loop, lightly touch the loop to a green colony and immerse it in the "+" tube. Using a new sterile loop, repeat for a white colony and immerse it in the "-" tube (it is very important to pick only a single colony). Spin the loop between your index finger and thumb to disperse the entire colony.
3. Cap the tubes and place them in the shaking incubator or on the shaking platform and culture overnight at 32 °C or 2 days at room temperature. Or cap the tubes and shake vigorously by hand. Place in the incubator horizontally at 32 °C for 24–48 hours. Remove and shake by hand periodically when possible.
Bacterial Concentration
1. Remove “+” liquid cultures from the shaker and observe with the UV light. Note any color differences between the two cultures. Label 2 microtubes with your group number. Transfer 1.5 ml of "+" liquid culture into each microtube. Spin the microtubes for 1 minute at 12000 rpm.
2. Pour out the supernatant and observe the pellet under UV light.
3. Add 100 µl of TE buffer to each tube. Resuspend the pellet thoroughly by rapidly pipetting up and down several times. Combine the samples into one tube (~ 200µl)
4. Add 20 µl of lysozyme to the resuspended bacterial pellet to initiate enzymatic digestion of the bacterial cell wall. Mix the contents gently by flicking the tube. Incubate at room temperature for 20 minutes. Observe the tube under the UV light.
5. Place the microtube in the -20oC freezer for 20 min. The freezing causes the bacteria to rupture completely.
6. Remove the microtube from the freezer and thaw using hand warmth.Place the tube in the centrifuge and pellet the insoluble bacterial debris by spinning for 5 minutes at 12000rpm.
7. After the 5 minute spin, immediately remove your tube from the centrifuge. Examine the tube with the UV light.
8. Transfer the supernatant into a new microtube labeled "+". This is the sample for HIC chromatography.
Bacterial Lysis
1. Remove the microtube from the freezer and thaw using hand warmth. Add 200µl (same volume) Binding buffer to mix with the samples.
Hydrophobic Interaction Chromatography (HIC)
1. Add 1 ml HIC matrix suspension to the empty column. Allow all of the liquid buffer to drain from the column (~3–5minutes).
2. Prepare the column by adding 1 ml of Equilibration buffer to the top of the column. Drain the buffer to the 1 ml mark on the column.
3. Label collection tubes 1, 2, 3, … and place the tubes in a rack. Place the column in collection tube 1. When the last of the buffer has reached the surface of the HIC matrix proceed to the next step below.
4. Carefully and gently load HIC sample onto the top of the column. Hold the pipette tip against the side of the column wall, just above the upper surface of the matrix and let the supernatant drip down the side of the column wall. Examine the column using a UV light. Note your observations. After it stops dripping transfer the column to collection tube 2.
5. Add 1 ml of Wash buffer and let the entire volume flow into the column. Examine the column using the UV light. Note your observations. After the column stops dripping, transfer it to tube 3.
6. Add 500 µl of TE Buffer and let the entire volume flow into the column. Examine the column using the UV light and note your observations.
7. Repeat step 6 two more times. Examine all collection tubes and note any differences in color between the tubes.
8. Transfer 500µl of the greenest sample into two microtubes each (labeled with your group number). These are the samples for gel electrophoresis next time.
HIC matrix regeneration
1. HIC matrix material is very expensive and we need to regenerate it so we can use it again. To regenerate the column after this experiment, add about 2-3 column volumes of 1.3 M Wash Buffer. Follow with 3-5 column volume of distilled water. Now column is ready for reused immediately. For long term storage, the matrix is stored in 10% Ethanol.

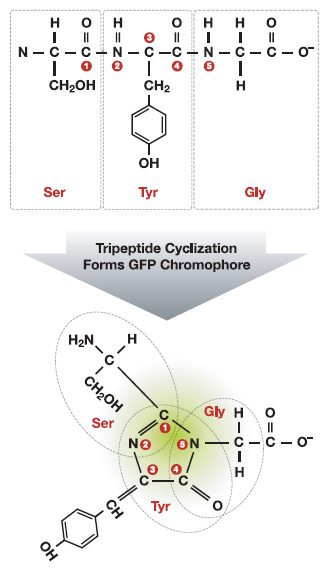
Figure 1. (Left). The tertiary structure of GFP is barrel-like, consisting of 11 beta sheets depicted as the ribbons and an internal chromophore of 3 adjacent amino acids, depicted as ball-and-stick. (Right) Cyclization of the tripeptide Ser-Tyr-Gly. The active chromophore of GFP is comprised of three adjacent amino acids in the primary polypeptide chain. The three amino acids are enzymatically converted to an active cyclic chromophore in vivo.
Laboratory write-up
1. What color was the bacterial pellet after centrifuging the intact cells? After centrifuging the lysed cells? What color were the supernatants? What does this tell you?
2. List your predictions and observations for the sample and what happens to the sample when the following buffers are added to the HIC column.
|
Collection tube No. |
Prediction |
Observations under UV (Column and collection tube) |
|
Tube 1 Sample in Binding Buffer |
||
|
Tube 2 Sample in Wash Buffer |
||
|
Tube 3 Sample in Elution Buffer |
3. Using the data table above, compare how your predictions matched up with your observations for each buffer.
a. Binding buffer
b. Wash buffer
c. Elution buffer
4. Based on your results, explain the roles or functions of these buffers.
a. Equilibration buffer
b. Binding buffer
c. Wash buffer
d. TE (Elution) buffer
5. Were you successful in isolating and purifying GFP from the cloned bacterial cells? Identify the evidence you have to support your answer.