
4.6: Example of Human Mutations
- Page ID
- 27250

– Cystic Fibrosis (CF) – autosmal recessive
Cystic fibrosis (CF) is one of many diseases that geneticists have shown to be caused by mutation of a single, well-characterized gene. Cystic fibrosis is the most common (1/2,500) life-limiting autosomal recessive disease among people of European heritage, with ~ 1 in 25 people being carriers. The frequency varies in different populations. Most of the deaths caused by CF are the result of lung disease, but many CF patients also suffer from other disorders including infertility and gastrointestinal disease. The disease is due to a mutation in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene, which was identified by Lap-chee Tsui’s group at the University of Toronto.
Epithelial tissues in some organs rely on the CFTR protein to transport ions (especially Cl-) across their cell membranes. The passage of ions through a six-sided channel is gated by another part of the CFTR protein, which binds to ATP. If there is insufficient activity of CFTR, an imbalance in ion concentration results, which disrupts the properties of the liquid layer that normally forms at the epithelial surface. In the lungs, this causes mucus to accumulate and can lead to infection. Defects in CFTR also affect pancreas, liver, intestines, and sweat glands, all of which need this ion transport. CFTR is also expressed at high levels in the salivary gland and bladder, but defects in CFTR function do not cause problems in these organs, probably because other ion transporters are able to compensate.
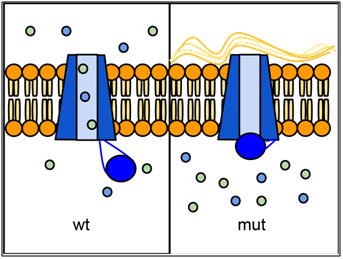
Over one thousand different mutant alleles of CFTR have been described. Any mutation that prevents CFTR from sufficiently transporting ions can lead to cystic fibrosis (CF). Worldwide, the most common CFTR allele among CF patients is called ΔF508 (delta-F508; or PHE508DEL), which is a deletion of three nucleotides that eliminates a phenylalanine from position 508 of the 1480 aa wild-type protein. Mutation ΔF508 causes CFTR to be folded improperly in the endoplasmic reticulum (ER), which prevents CFTR from reaching the cell membrane. ΔF508 accounts for approximately 70% of CF cases in North America, with ~1/25 people of European descent being carriers. The high frequency of the ΔF508 allele has led to speculation that it may confer some selective advantage to heterozygotes, perhaps by reducing dehydration during cholera epidemics, or by reducing susceptibility to certain pathogens that bind to epithelial membranes.
CFTR is also notable because it is one of the well-characterized genetic diseases for which a drug has been developed that compensates for the effects of a specific mutation. The drug, Kalydeco, was approved by the FDA and Health Canada in 2012, decades after the CFTR gene was first mapped to DNA markers (in 1985) and cloned (in 1989). Kalydeco is effective on only some CFTR mutations, most notably G551D (i.e. where glycine is substituted by aspartic acid at position 551 of the protein; GLY551ASP). This mutation is found in less than 5% of CF patients. The G551D mutation affects the ability of ATP to bind to CFTR and open the channel it for transport. Kalydeco compensates for mutation by binding to CFTR and holding it in an open conformation. Kalydeco is expected to cost approximately $250,000 per patient per year.