
17.5B: Basic Techniques in Protein Analysis
- Page ID
- 13389

- Describe the techniques used in proteomics to analyze proteins
Basic Techniques in Protein Analysis
The ultimate goal of proteomics is to identify or compare the proteins expressed in a given genome under specific conditions, study the interactions between the proteins, and use the information to predict cell behavior or develop drug targets. Just as the genome is analyzed using the basic technique of DNA sequencing, proteomics requires techniques for protein analysis. The basic technique for protein analysis, analogous to DNA sequencing, is mass spectrometry.

Mass Spectrometry
Mass spectrometry is used to identify and determine the characteristics of a molecule. It is a technique in which gas phase molecules are ionized and their mass-to-charge ratio is measured by observing acceleration differences of ions when an electric field is applied. Lighter ions will accelerate faster and be detected first. If the mass is measured with precision, then the composition of the molecule can be identified. In the case of proteins, the sequence can be identified. The challenge of techniques used for proteomic analyses is the difficulty in detecting small quantities of proteins, but advances in spectrometry have allowed researchers to analyze very small samples of protein. Variations in protein expression in diseased states, however, can be difficult to discern. Proteins are naturally-unstable molecules, which makes proteomic analysis much more difficult than genomic analysis.
X-ray crystallography and Nuclear Magnetic Resonance
X-ray crystallography enables scientists to determine the three-dimensional structure of a protein crystal at atomic resolution. Crystallographers aim high-powered X-rays at a tiny crystal containing trillions of identical molecules. The crystal scatters the X-rays onto an electronic detector that is the same type used to capture images in a digital camera. After each blast of X-rays, lasting from a few seconds to several hours, the researchers precisely rotate the crystal by entering its desired orientation into the computer that controls the X-ray apparatus. This enables the scientists to capture in three dimensions how the crystal scatters, or diffracts, X-rays. The intensity of each diffracted ray is fed into a computer, which uses a mathematical equation to calculate the position of every atom in the crystallized molecule. The result is a three-dimensional digital image of the molecule.
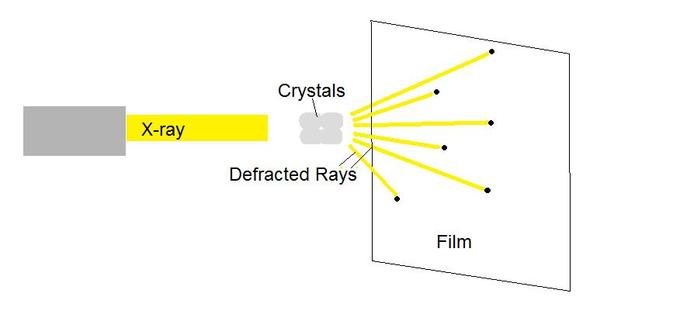
Another protein imaging technique, nuclear magnetic resonance (NMR), uses the magnetic properties of atoms to determine the three-dimensional structure of proteins. NMR spectroscopy is unique in being able to reveal the atomic structure of macromolecules in solution, provided that highly-concentrated solution can be obtained. This technique depends on the fact that certain atomic nuclei are intrinsically magnetic. The chemical shift of nuclei depends on their local environment. The spins of neighboring nuclei interact with each other in ways that provide definitive structural information that can be used to determine complete three-dimensional structures of proteins.
Protein Microarrays and Two- Hybrid Screening
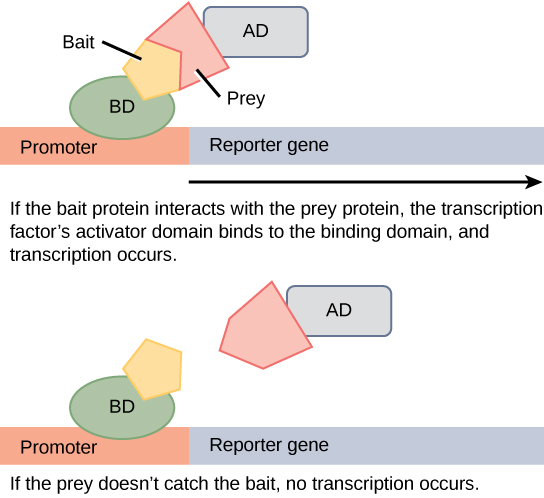
Protein microarrays have also been used to study interactions between proteins. These are large-scale adaptations of the basic two-hybrid screen. The premise behind the two-hybrid screen is that most eukaryotic transcription factors have modular activating and binding domains that can still activate transcription even when split into two separate fragments, as long as the fragments are brought within close proximity to each other. Generally, the transcription factor is split into a DNA-binding domain (BD) and an activation domain (AD). One protein of interest is genetically fused to the BD and another protein is fused to the AD. If the two proteins of interest bind each other, then the BD and AD will also come together and activate a reporter gene that signals interaction of the two hybrid proteins.
Western Blot
The western blot, or protein immunoblot, is a technique that combines protein electrophoresis and antibodies to detect proteins in a sample. A western blot is fairly quick and simple compared to the above techniques and, thus, can serve as an assay to validate results from other experiments. The protein sample is first separated by gel electrophoresis, then transferred to a nitrocellulose or other type of membrane, and finally stained with a primary antibody that specifically binds the protein of interest. A fluorescent or radioactive-labeled secondary antibody binds to the primary antibody and provides a means of detection via either photography or x-ray film, respectively.
Key Points
- Mass Spectrometry is a technique that is useful for determining the size of a protein or protein complex.
- X-ray crystallography and NMR are techniques useful for determining the 3-D structure of a protein or protein complex.
- Protein microarrays are useful for determining protein-protein interactions.
Key Terms
- microarray: any of several devices containing a two-dimensional array of small quantities of biological material used for various types of assays
- reporter gene: a gene that researchers attach to a regulatory sequence of another gene of interest and whose product is easily identifiable in assays