
2.3: Maximum Likelihood
- Page ID
- 21581

Section 2.3a: What is a likelihood?
Since all of the approaches described in the remainer of this chapter involve calculating likelihoods, I will first briefly describe this concept. A good general review of likelihood is Edwards (1992). Likelihood is defined as the probability, given a model and a set of parameter values, of obtaining a particular set of data. That is, given a mathematical description of the world, what is the probability that we would see the actual data that we have collected?
To calculate a likelihood, we have to consider a particular model that may have generated the data. That model will almost always have parameter values that need to be specified. We can refer to this specified model (with particular parameter values) as a hypothesis, H. The likelihood is then:
\[L(H|D)=Pr(D|H) \label{2.1}\]
Here, L and Pr stand for likelihood and probability, D for the data, and H for the hypothesis, which again includes both the model being considered and a set of parameter values. The | symbol stands for “given,” so equation 2.1 can be read as “the likelihood of the hypothesis given the data is equal to the probability of the data given the hypothesis.” In other words, the likelihood represents the probability under a given model and parameter values that we would obtain the data that we actually see.
For any given model, using different parameter values will generally change the likelihood. As you might guess, we favor parameter values that give us the highest probability of obtaining the data that we see. One way to estimate parameters from data, then, is by finding the parameter values that maximize the likelihood; that is, the parameter values that give the highest likelihood, and the highest probability of obtaining the data. These estimates are then referred to as maximum likelihood (ML) estimates. In an ML framework, we suppose that the hypothesis that has the best fit to the data is the one that has the highest probability of having generated that data.
For the example above, we need to calculate the likelihood as the probability of obtaining heads 63 out of 100 lizard flips, given some model of lizard flipping. In general, we can write the likelihood for any combination of H “successes” (flips that give heads) out of n trials. We will also have one parameter, pH, which will represent the probability of “success,” that is, the probability that any one flip comes up heads. We can calculate the likelihood of our data using the binomial theorem:
\[ L(H|D)=Pr(D|p)= {n \choose H} p_H^H (1-p_H)^{n-H} \label{2.2} \]
In the example given, n = 100 and H = 63, so:
\[ L(H|D)= {100 \choose 63} p_H^{63} (1-p_H)^{37} \label{2.3} \]
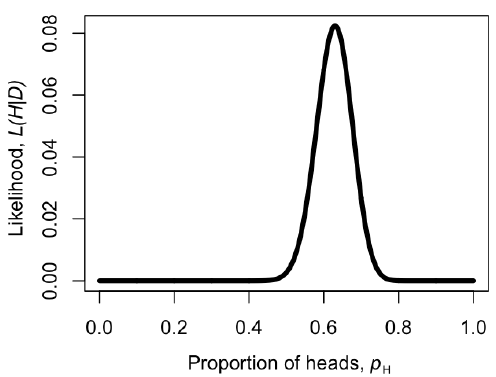
We can make a plot of the likelihood, L, as a function of pH (Figure 2.2). When we do this, we see that the maximum likelihood value of pH, which we can call $\hat{p}_H$, is at $\hat{p}_H = 0.63$. This is the “brute force” approach to finding the maximum likelihood: try many different values of the parameters and pick the one with the highest likelihood. We can do this much more efficiently using numerical methods as described in later chapters in this book.
We could also have obtained the maximum likelihood estimate for pH through differentiation. This problem is much easier if we work with the ln-likelihood rather than the likelihood itself (note that whatever value of pH that maximizes the likelihood will also maximize the ln-likelihood, because the log function is strictly increasing). So:
\[ \ln{L} = \ln{n \choose H} + H \ln{p_H}+ (n-H)\ln{(1-p_H)} \label{2.4} \]
Note that the natural log (ln) transformation changes our equation from a power function to a linear function that is easy to solve. We can differentiate:
\[ \frac{d \ln{L}}{dp_H} = \frac{H}{p_H} - \frac{(n-H)}{(1-p_H)}\label{2.5} \]
The maximum of the likelihood represents a peak, which we can find by setting the derivative $\frac{d \ln{L}}{dp_H}$ to zero. We then find the value of pH that solves that equation, which will be our estimate $\hat{p}_H$. So we have:
\[ \begin{array}{lcl} \frac{H}{\hat{p}_H} - \frac{n-H}{1-\hat{p}_H} & = & 0\\ \frac{H}{\hat{p}_H} & = & \frac{n-H}{1-\hat{p}_H}\\ H (1-\hat{p}_H) & = & \hat{p}_H (n-H)\\ H-H\hat{p}_H & = & n\hat{p}_H-H\hat{p}_H\\ H & = & n\hat{p}_H\\ \hat{p}_H &=& H / n\\ \end{array} \label{2.6}\]
Notice that, for our simple example, H/n = 63/100 = 0.63, which is exactly equal to the maximum likelihood from figure 2.2.
Maximum likelihood estimates have many desirable statistical properties. It is worth noting, however, that they will not always return accurate parameter estimates, even when the data is generated under the actual model we are considering. In fact, ML parameters can sometimes be biased. To understand what this means, we need to formally introduce two new concepts: bias and precision. Imagine that we were to simulate datasets under some model A with parameter a. For each simulation, we then used ML to estimate the parameter $\hat{a}$ for the simulated data. The precision of our ML estimate tells us how different, on average, each of our estimated parameters $\hat{a}_i$ are from one another. Precise estimates are estimated with less uncertainty. Bias, on the other hand, measures how close our estimates $\hat{a}_i$ are to the true value a. If our ML parameter estimate is biased, then the average of the $\hat{a}_i$ will differ from the true value a. It is not uncommon for ML estimates to be biased in a way that depends on sample size, so that the estimates get closer to the truth as sample size increases, but can be quite far off in a particular direction when the number of data points is small compared to the number of parameters being estimated.
In our example of lizard flipping, we estimated a parameter value of $\hat{p}_H = 0.63$. For the particular case of estimating the parameter of a binomial distribution, our ML estimate is known to be unbiased. And this estimate is different from 0.5 – which was our expectation under the null hypothesis. So is this lizard fair? Or, alternatively, can we reject the null hypothesis that pH = 0.5? To evaluate this, we need to use model selection.
Section 2.3b: The likelihood ratio test
Model selection involves comparing a set of potential models and using some criterion to select the one that provides the “best” explanation of the data. Different approaches define “best” in different ways. I will first discuss the simplest, but also the most limited, of these techniques, the likelihood ratio test. Likelihood ratio tests can only be used in one particular situation: to compare two models where one of the models is a special case of the other. This means that model A is exactly equivalent to the more complex model B with parameters restricted to certain values. We can always identify the simpler model as the model with fewer parameters. For example, perhaps model B has parameters x, y, and z that can take on any values. Model A is the same as model B but with parameter z fixed at 0. That is, A is the special case of B when parameter z = 0. This is sometimes described as model A is nested within model B, since every possible version of model A is equal to a certain case of model B, but model B also includes more possibilities.
For likelihood ratio tests, the null hypothesis is always the simpler of the two models. We compare the data to what we would expect if the simpler (null) model were correct.
For example, consider again our example of flipping a lizard. One model is that the lizard is “fair:” that is, that the probability of heads is equal to 1/2. A different model might be that the probability of heads is some other value p, which could be 1/2, 1/3, or any other value between 0 and 1. Here, the latter (complex) model has one additional parameter, pH, compared to the former (simple) model; the simple model is a special case of the complex model when pH = 1/2.
For such nested models, one can calculate the likelihood ratio test statistic as
\[ \Delta = 2 \cdot \ln{\frac{L_1}{L_2}} = 2 \cdot (\ln{L_1}-\ln{L_2}) \label{2.7}\]
Here, Δ is the likelihood ratio test statistic, L2 the likelihood of the more complex (parameter rich) model, and L1 the likelihood of the simpler model. Since the models are nested, the likelihood of the complex model will always be greater than or equal to the likelihood of the simple model. This is a direct consequence of the fact that the models are nested. If we find a particular likelihood for the simpler model, we can always find a likelihood equal to that for the complex model by setting the parameters so that the complex model is equivalent to the simple model. So the maximum likelihood for the complex model will either be that value, or some higher value that we can find through searching the parameter space. This means that the test statistic Δ will never be negative. In fact, if you ever obtain a negative likelihood ratio test statistic, something has gone wrong – either your calculations are wrong, or you have not actually found ML solutions, or the models are not actually nested.
To carry out a statistical test comparing the two models, we compare the test statistic Δ to its expectation under the null hypothesis. When sample sizes are large, the null distribution of the likelihood ratio test statistic follows a chi-squared (χ2) distribution with degrees of freedom equal to the difference in the number of parameters between the two models. This means that if the simpler hypothesis were true, and one carried out this test many times on large independent datasets, the test statistic would approximately follow this χ2 distribution. To reject the simpler (null) model, then, one compares the test statistic with a critical value derived from the appropriate χ2 distribution. If the test statistic is larger than the critical value, one rejects the null hypothesis. Otherwise, we fail to reject the null hypothesis. In this case, we only need to consider one tail of the χ2 test, as every deviation from the null model will push us towards higher Δ values and towards the right tail of the distribution.
For the lizard flip example above, we can calculate the ln-likelihood under a hypothesis of pH = 0.5 as:
\[ \begin{array}{lcl} \ln{L_1} &=& \ln{\left(\frac{100}{63}\right)} + 63 \cdot \ln{0.5} + (100-63) \cdot \ln{(1-0.5)} \nonumber \\ \ln{L_1} &=& -5.92\nonumber\\ \end{array} \label{2.8}\]
We can compare this to the likelihood of our maximum-likelihood estimate :
\[ \begin{array}{lcl} \ln{L_2} &=& \ln{\left(\frac{100}{63}\right)} + 63 \cdot \ln{0.63} + (100-63) \cdot \ln{(1-0.63)} \nonumber \\ \ln{L_2} &=& -2.50\nonumber \end{array} \label{2.9}\]
We then calculate the likelihood ratio test statistic:
\[ \begin{array}{lcl} \Delta &=& 2 \cdot (\ln{L_2}-\ln{L_1}) \nonumber \\ \Delta &=& 2 \cdot (-2.50 - -5.92) \nonumber \\ \Delta &=& 6.84\nonumber \end{array} \label{2.10}\]
If we compare this to a χ2 distribution with one d.f., we find that P = 0.009. Because this P-value is less than the threshold of 0.05, we reject the null hypothesis, and support the alternative. We conclude that this is not a fair lizard. As you might expect, this result is consistent with our answer from the binomial test in the previous section. However, the approaches are mathematically different, so the two P-values are not identical.
Although described above in terms of two competing hypotheses, likelihood ratio tests can be applied to more complex situations with more than two competing models. For example, if all of the models form a sequence of increasing complexity, with each model a special case of the next more complex model, one can compare each pair of hypotheses in sequence, stopping the first time the test statistic is non-significant. Alternatively, in some cases, hypotheses can be placed in a bifurcating choice tree, and one can proceed from simple to complex models down a particular path of paired comparisons of nested models. This approach is commonly used to select models of DNA sequence evolution (Posada and Crandall 1998).
Section 2.3c: The Akaike Information Criterion (AIC)
You might have noticed that the likelihood ratio test described above has some limitations. Especially for models involving more than one parameter, approaches based on likelihood ratio tests can only do so much. For example, one can compare a series of models, some of which are nested within others, using an ordered series of likelihood ratio tests. However, results will often depend strongly on the order in which tests are carried out. Furthermore, often we want to compare models that are not nested, as required by likelihood ratio tests. For these reasons, another approach, based on the Akaike Information Criterion (AIC), can be useful.
The AIC value for a particular model is a simple function of the likelihood L and the number of parameters k:
\[AIC = 2k − 2\ln L \label{2.11}\]
This function balances the likelihood of the model and the number of parameters estimated in the process of fitting the model to the data. One can think of the AIC criterion as identifying the model that provides the most efficient way to describe patterns in the data with few parameters. However, this shorthand description of AIC does not capture the actual mathematical and philosophical justification for equation (2.11). In fact, this equation is not arbitrary; instead, its exact trade-off between parameter numbers and log-likelihood difference comes from information theory (for more information, see Burnham and Anderson 2003, Akaike (1998)).
The AIC equation (2.11) above is only valid for quite large sample sizes relative to the number of parameters being estimated (for n samples and k parameters, n/k > 40). Most empirical data sets include fewer than 40 independent data points per parameter, so a small sample size correction should be employed:
\[ AIC_C = AIC + \frac{2k(k+1)}{n-k-1} \label{2.12}\]
This correction penalizes models that have small sample sizes relative to the number of parameters; that is, models where there are nearly as many parameters as data points. As noted by Burnham and Anderson (2003), this correction has little effect if sample sizes are large, and so provides a robust way to correct for possible bias in data sets of any size. I recommend always using the small sample size correction when calculating AIC values.
To select among models, one can then compare their AICc scores, and choose the model with the smallest value. It is easier to make comparisons in AICc scores between models by calculating the difference, ΔAICc. For example, if you are comparing a set of models, you can calculate ΔAICc for model i as:
\[ΔAIC_{c_i} = AIC_{c_i} − AIC_{c_{min}} \label{2.13}\]
where AICci is the AICc score for model i and AICcmin is the minimum AICc score across all of the models.
As a broad rule of thumb for comparing AIC values, any model with a ΔAICci of less than four is roughly equivalent to the model with the lowest AICc value. Models with ΔAICci between 4 and 8 have little support in the data, while any model with a ΔAICci greater than 10 can safely be ignored.
Additionally, one can calculate the relative support for each model using Akaike weights. The weight for model i compared to a set of competing models is calculated as:
\[ w_i = \frac{e^{-\Delta AIC_{c_i}/2}}{\sum_i{e^{-\Delta AIC_{c_i}/2}}} \label{2.14} \]
The weights for all models under consideration sum to 1, so the wi for each model can be viewed as an estimate of the level of support for that model in the data compared to the other models being considered.
Returning to our example of lizard flipping, we can calculate AICc scores for our two models as follows:
\[ \begin{array}{lcl} AIC_1 &=& 2 k_1 - 2 ln{L_1} = 2 \cdot 0 - 2 \cdot -5.92 \\\ AIC_1 &=& 11.8 \\\ AIC_2 &=& 2 k_2 - 2 ln{L_2} = 2 \cdot 1 - 2 \cdot -2.50 \\\ AIC_2 &=& 7.0 \\\ \end{array} \label{2.15} \]
Our example is a bit unusual in that model one has no estimated parameters; this happens sometimes but is not typical for biological applications. We can correct these values for our sample size, which in this case is n = 100 lizard flips:
\[ \begin{array}{lcl} AIC_{c_1} &=& AIC_1 + \frac{2 k_1 (k_1 + 1)}{n - k_1 - 1} \\\ AIC_{c_1} &=& 11.8 + \frac{2 \cdot 0 (0 + 1)}{100-0-1} \\\ AIC_{c_1} &=& 11.8 \\\ AIC_{c_2} &=& AIC_2 + \frac{2 k_2 (k_2 + 1)}{n - k_2 - 1} \\\ AIC_{c_2} &=& 7.0 + \frac{2 \cdot 1 (1 + 1)}{100-1-1} \\\ AIC_{c_2} &=& 7.0 \\\ \end{array} \label{2.16} \]
Notice that, in this particular case, the correction did not affect our AIC values, at least to one decimal place. This is because the sample size is large relative to the number of parameters. Note that model 2 has the smallest AICc score and is thus the model that is best supported by the data. Noting this, we can now convert these AICc scores to a relative scale:
\[ \begin{array}{lcl} \Delta AIC_{c_1} &=& AIC_{c_1}-AIC{c_{min}} \\\ &=& 11.8-7.0 \\\ &=& 4.8 \\\ \end{array} \label{2.17} \]
\[ \begin{array}{lcl} \Delta AIC_{c_2} &=& AIC_{c_2}-AIC{c_{min}} \\\ &=& 7.0-7.0 \\\ &=& 0 \\\ \end{array} \]
Note that the ΔAICci for model 1 is greater than four, suggesting that this model (the “fair” lizard) has little support in the data. This is again consistent with all of the results that we've obtained so far using both the binomial test and the likelihood ratio test. Finally, we can use the relative AICc scores to calculate Akaike weights:
\[ \begin{array}{lcl} \sum_i{e^{-\Delta_i/2}} &=& e^{-\Delta_1/2} + e^{-\Delta_2/2} \\\ &=& e^{-4.8/2} + e^{-0/2} \\\ &=& 0.09 + 1 \\\ &=& 1.09 \\\ \end{array} \label{2.18}\]
\[ \begin{array}{lcl} w_1 &=& \frac{e^{-\Delta AIC_{c_1}/2}}{\sum_i{e^{-\Delta AIC_{c_i}/2}}} \\\ &=& \frac{0.09}{1.09} \\\ &=& 0.08 \end{array} \]
\[ \begin{array}{lcl} w_2 &=& \frac{e^{-\Delta AIC_{c_2}/2}}{\sum_i{e^{-\Delta AIC_{c_i}/2}}} \\\ &=& \frac{1.00}{1.09} \\\ &=& 0.92 \end{array} \]
Our results are again consistent with the results of the likelihood ratio test. The relative likelihood of an unfair lizard is 0.92, and we can be quite confident that our lizard is not a fair flipper.
AIC weights are also useful for another purpose: we can use them to get model-averaged parameter estimates. These are parameter estimates that are combined across different models proportional to the support for those models. As a thought example, imagine that we are considering two models, A and B, for a particular dataset. Both model A and model B have the same parameter p, and this is the parameter we are particularly interested in. In other words, we do not know which model is the best model for our data, but what we really need is a good estimate of p. We can do that using model averaging. If model A has a high AIC weight, then the model-averaged parameter estimate for p will be very close to our estimate of p under model A; however, if both models have about equal support then the parameter estimate will be close to the average of the two different estimates. Model averaging can be very useful in cases where there is a lot of uncertainty in model choice for models that share parameters of interest. Sometimes the models themselves are not of interest, but need to be considered as possibilities; in this case, model averaging lets us estimate parameters in a way that is not as strongly dependent on our choice of models.