
3.3: Changes in Protein Shape Can Cause Disease
- Page ID
- 16424

While the conformation of a protein determines its biological function, an allosteric change (change in shape) can moderate or disrupt its function. Under normal circumstances, cells use changes in protein shape to regulate metabolism. Such allosteric regulation is well documented in familiar biochemical pathways such as glycolysis and is discussed in more detail elsewhere. Less well understood is how (or why) is why conformational change in some proteins cells has devastating effects.
A. Sickle Cell Anemia
Mutant genes for globins can result in hemoglobin disorders characterized by inefficient oxygen delivery by blood. In the 1940s, the British biochemist J.B.S. Haldane made a correlation between southern African regions with high incidences hemoglobin disorders and malaria, suggesting that heterozygous individuals (i.e., those that had only one copy of a mutant hemoglobin gene), were somehow protected from malaria. Another well-known example of a hemoglobin disorder is sickle cell anemia, caused by a single base change in the gene for human b-hemoglobin, one of the polypeptides in hemoglobin. Since red blood cells are rich in hemoglobin, sickling hemoglobins can cause the cells themselves to become sickle-shape. Sickled cells disrupt capillary flow and oxygen delivery, resulting in the symptoms of anemia. Sickle cell anemia originated in Africa and spread to the United States during the slave trade. Once out of Africa and regions where malaria was epidemic, the mutation was of no value, and was just a source of disease. Individuals heterozygous for the sickle cell mutation have sickle cell trait and are generally unaffected because at least some of their hemoglobin is normal. Homozygous individuals make only the sickle cell variant of b-hemoglobin; they will suffer more frequent and severe suffer episodes of the disease. Stressors that can trigger sickling include infection or dehydration. Compare normal red blood cells to a sickle cell below.
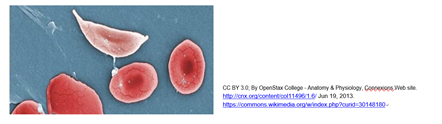
The sickle cell gene affects perhaps more than 100 million people worldwide, including 8-10% of African Americans. For more demographic information, see Sickle Cell Trait Demographics Article and Sickle Cell Data from the CDC. In Africa, heterozygotes with sickle cell trait are protected from malaria, confirming Haldane’s hypothesis. But patients homozygous for the b-hemoglobin mutation derive little benefit from its anti- malarial effects.
In the meantime, despite a 33% reduction in cases of malaria, the disease (caused by a mosquito-borne parasite) still threatens half of the people on the planet, causing over a half-million deaths per year. There are treatments other than mosquito nets and killing mosquitos, but at this time, there is still no preventive vaccine.
B. The Mis-Folding of Prions and Alzheimer’s Disease
1. The Prion Protein
When first discovered, prion proteins seemed to behave as infectious agents that could reproduce without DNA or other nucleic acid. As you can imagine, this highly unorthodox and novel hereditary mechanism generated its share of controversy. Read about research on the cellular PrPc prion protein at en.Wikipedia.org/wiki/Prion.
Of course, prions turned out not to be reproductive agents of infection after all. Recent studies of prions have revealed several normal prion protein functions such as roles in memory formation in mice and in sporulation in yeast (Check out Prion Proteins May Play a Role in Memory Formation). A mutant version of the prion protein (PrPSc) is able to mis-fold, assuming an abnormal shape. The deformed PrPSc can then induce abnormal folding even normal PrPc! These events, illustrated below, result in the formation of so-called amyloid plaques.

In their abnormally folded state, prions have been associated Alzheimer’s Disease (which affects about 5.5 million Americans, as well as with Mad Cow disease and Creutzfeldt-Jakob-Disease (mad cow disease in humans), as well as Scrapie in sheep, among others. We are beginning to understand that the role of prion proteins in Alzheimer’s Disease is less causal and somewhat indirect.
2. The amyloid beta (Ab) peptide
The post-mortem brains of patients that suffered Alzheimer’s disease exhibit characteristic extracellular amyloid plaques composed largely of the amyloid beta (Ab) peptide. Enzymatic cleavage of the APP protein (amyloid precursor protein) generates extracellular 39-43 amino acid Ab peptides. Under normal conditions, excess Ab peptides are themselves digested.
Unregulated Ab peptide formation however, leads to the formation of beta amyloid plaques seen in Alzheimer’s disease.as illustrated below.

The scissors in the illustration represent two enzymes that digest the APP. Prion proteins are not a proximal cause of Alzheimer’s Disease, but may have a role in initiating events that lead to it. Normal prion protein (PrPc), itself a membrane receptor, is thought to bind Ab peptides, effectively preventing their aggregation into plaques. An experimental reduction of PrPc was shown to increases extracellular Ab peptides. Presumably prion protein aggregation induced by mutant PrP (PrPsc) prevents prion proteins from binding to Ab peptide, leading to its accumulation and ultimately to amyloid plaque formation and neurodegeneration.
3. The Tau protein
A protein called tau is also associated with Alzheimer’s Disease. Misshapen tau accumulating in neurofibrillary tangles in hippocampus brain neurons may be a more immediate cause of the neuronal disfunction associated with the disease. In normal neurons, a Microtubule-Associated Protein Tau (MAP-T) is phosphory- lated and then binds to, and stabilizes microtubules. But when neuronal tau becomes hyper-phosphorylated, its conformation changes. No longer stabilized, the microtubules disassemble and the deformed tau proteins form neurofibrillary tangles. Immunostaining of hippocampal neurons with antibodies to tau protein localizes the neurofibrillary tangles as seen in the micrograph below.

The formation of neurofibrillary tau protein tangles in a diseased neuron is compared to normal neurons in the illustration below.

The” tangles clumps of tau proteins” in this illustration are what stain deep purple in the micrograph of immunostained neurons in the light micrograph.
There is no cure for Alzheimer’s disease, although treatments with cholinesterase inhibitors seem to slow its advancement. For example, the drug Aricept inhibits acetylcholine breakdown by acetylcholinesterase, thereby enhancing cholinergic neurotransmission, which may in turn prolong brain neural function. Unfortunately, there is as yet no treatment to restore lost memories and the significant cognitive decline associated with Alzheimer’s disease. Perhaps more promising in this respect, the recent development of a blood test may detect people at risk for Alzheimer’s disease. As it happens, Aβ molecules escape into the blood stream as much as 8 years before Alzheimer’s symptoms appear. The prospect of early Aβ detection has raised hopes that new therapies might be on the horizon. For a brief review, see Early Detection of Alzheimer's Disease.
C. Some Relatives of Alzheimer’s Disease
Some of the same protein abnormalities that are seen in Alzheimer’s disease also characterize other neurodegenerative diseases as well as traumatic brain damage, as discussed below.
1. Chronic Traumatic Encephalopathy
An abnormal accumulation of tau protein is diagnostic of CTE (Chronic Traumatic Encephalopathy). In the early 20th century, disoriented boxers staggering about after a fight were called ‘punch drunk’, suffering from dementia pugilistica. We now know they suffered from CTE, as do other athletes exposed to repetitive mild-to- severe brain trauma, such as football players. Immunostaining of whole brains and brain tissue from autopsied CTE patients with antibodies to tau protein show accumulations of abnormal tau proteins and tau neurofibrillary tangles very much like those found in of Alzheimer’s patients. Many National Football League and other football players have been diagnosed post-mortem with CTE, and many still living show signs of degenerative cognition and behavior consistent with CTE (see a List of_NFL_players_with_chronic_traumatic_encephalopathy to see how many!
2. Parkinson’s Disease
This is yet another example of a neurodegenerative disease that results when a single protein changes shape in brain cells. Though not characterized as plaques, aggregates can form in brain cells when the protein alpha-synuclein undergoes anomalous conformational change. The change results in MSA (Multiple System Atrophy) or Parkinson’s Disease (click Synuclein Allostery and Aggregation in Parkinson's Disease to read more details about this recent research). Much of the high-resolution electron microscopy that reveals protein structure and that can capture conformational changes we now recognize, comes from the work of Jacques Dubochet, Joachim Frank and Richard Henderson who received the 2017 Chemistry Nobel Prize for Chemistry for developing and refining cryo-electron microscopy for biomolecular imaging (see 2017 Nobel Prize for Chemistry for more).