
5.2: Good Clinical Practices (GCP)
- Page ID
- 39499

The government has an interest in protecting the public from defective products and drugs. Therefore, companies must demonstrate their effectiveness and safety before mass distribution. However, the only way they can actively do this is by having human subjects test out their products.
If a potential new product appears safe in animal studies, then a plan is created to investigate the product in clinical trials using human volunteers. The company submits its plan to the FDA in an IND. The IND application includes a description of the product, the results of animal tests, and the plans for further testing. The FDA then decides whether the company’s materials are sufficiently complete that the company can begin testing the product in humans.
Explore!
Explore more about Good Clinical Practices & Clinical Trials:
- http://fda.yorkcast.com/webcast/Play/477af877491747379c36c4ab1c7421b9
- Watch this video on clinical trials youtu.be/pm1igf85uoA
Good Clinical Practices (GCPs) apply to the performance of clinical trials of drug safety and efficacy in human subjects. GCPs aim to protect the rights and safety of human subjects and ensure the scientific quality of the studies. Clinical trials are conducted in stages, and each stage must be successful before continuing to the next phase. Good Clinical Practices (GCPs) are a similar set of standards that apply to human subjects of clinical trials and experiments.
Regulatory History of GCPs:
- The Nuremberg Code lists ten basic moral, ethical, and legal principles outlining medical research established in response to the Nuremberg doctor's trials in 1946. This tribunal launched criminal proceedings against physicians for crimes against humanity in WWI.
- In 1964, the World Medical Association established ethical guidelines for biomedical research in humans called the Declaration of Helsinki. These guidelines include essential codes of conduct, including areas involving informed consent, confidentiality, research protocol review, the risk versus benefit analysis, publication and data access to the scientific community, and the importance of the subject’s health over the interest of study.
- The Belmont Report (1979), established principles of ethical research emphasizing respect for persons, beneficence, and justice, leading to the Common Rule in 1981.
- By the 1980s, it became apparent that representative populations are needed in clinical trials - factors that may influence the effectiveness and side effects of drugs include age (children, older patients), sex, and ethnicity.
- In 1989, the FDA issued guidelines asking manufacturers to determine whether a drug is likely to have significant use in older people.
- In 1993, the FDA issued the Gender Guideline, which called for assessments of medication responses in both sexes
- In 1998, the FDA required that a marketing application analyzes data on safety and effectiveness by age, gender, and race, known as the Demographic Rule.
- In 2002, the Best Pharmaceuticals for Children Act was passed to improve the safety and effectiveness of medicines for children.
- In 2003, the FDA was given clear authority under the Pediatric Research Equity Act to require drug sponsors to conduct clinical research into pediatric applications for new drugs.
Explore!
Perform cursory internet research on Tuskegee Syphilis Study (1932-1972). Summarize why this incident would cause outrage and a public apology by a US President? In what ways did this violate the Declaration of Helsinki? What was the regulatory response? (Meaning, what law was passed?)
What Are Good Clinical Practices?
Although there is no regulation specifically entitled “Good Clinical Practice,” there are several regulations, which govern the conduct of clinical trials.
- Volunteers participating in a clinical study must be able to give informed consent. This means educating each potential subject on the treatment they are to receive as a part of the study as well as any risks that may be associated with their participation. FDA regulations entitled “Protections of Human Subjects” (21 CFR 50) set forth the requirements for informed consent.
- Clinical trials must be reviewed by a committee independent of the study sponsor called an Institutional Review Board (IRB) (21 CFR 50). The regulations specify the organization and personnel who make up this board, as well as the records and reports that are to be kept.
- 21 CFR 312 Subpart D outlines the responsibilities of trial sponsors and investigators during a trial. Additionally, the FDA “Guideline for the Monitoring of Clinical Investigations” explains monitoring and documentation.
Ethics of Clinical Studies
Many believe that ‘informed consent’ is all that is required to satisfy ethical concerns for clinical studies. It is far more complex than that. In addition to Informed consent, one must consider Social and clinical value, Scientific validity, Fair subject selection, Favorable risk-benefit ratio, Independent review, and Respect for potential and enrolled subjects. https://clinicalcenter.nih.gov/recruit/ethics.html
The goal of clinical research is to develop generalizable knowledge that improves human health or increases understanding of human biology. People who participate in clinical research make it possible to secure that knowledge. The path to finding out if a new drug or treatment is safe or effective, for example, is to test it on patient volunteers. However, by placing some people at risk of harm for the good of others, clinical research has the potential to exploit patient volunteers. The purpose of ethical guidelines is both to protect patient volunteers and to preserve the integrity of the science.
The ethical guidelines in place today were primarily a response to past abuses, the most notorious of which in America was an experiment in Tuskegee, Alabama, in which treatment was withheld from 400 African American men with syphilis so that scientists could study the course of the disease. Various ethical guidelines were developed in the 20th century in response to such studies.
The Belmont Report
There are many guidelines in addition to rules and regulations that govern clinical study ethics. Some of the more influential ones include The Nuremberg Code (1947), Declaration of Helsinki (2000), Belmont Report (1979), CIOMS (2002), and US Common Rule (1991). Read the Belmont Report here: https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/index.html
Informed Consent
Any patient participating in a clinical study must do so under informed consent. Some exceptions to this rule include military operations or public health emergencies. Informed consent to the FDA does not just include patient authorization but an exchange of information between the subject and the individual obtaining this approval. The subject must have enough information about the study to make an informed decision about their participation in the study. Informed consent is outlined in the Informed Consent Form (ICF), allows the subject time to reflect, and has the information available to do so, and therefore, the ICF is submitted to the FDA for review.
Institutional Review Board (IRB)
A company must also get approval from an Institutional Review Board (IRB) to perform human testing. The IRB is a group responsible for protecting the rights, safety, and wellbeing of human subjects. It is typically composed of a minimum of five, gender-diverse members; at least one science and one non-science member. The IRB general standards are covered and described in 21 CFR Part 56. FDA has a comprehensive list of regulations that govern Clinical Studies (Clinicalstudies.gov). International GCP guidance documents, which the FDA has collaborated, and links to other sites relevant to the conduct of clinical trials, both nationally and internationally are also found here.
Bioresearch Monitoring
The overarching goals of the FDA's bioresearch monitoring (BIMO) program are to protect the rights, safety, and welfare of subjects involved in FDA-regulated clinical trials; to determine the accuracy and reliability of clinical trial data submitted to FDA; and to assess compliance with FDA's regulations governing the conduct of clinical trials, including those for informed consent and ethical review. The BIMO program performs on-site inspections of both clinical and nonclinical studies performed to support research and marketing applications/submissions to the agency.
Office of Good Clinical Practice Mission Statement
The Office of Good Clinical Practice is the focal point within FDA for Good Clinical Practice (GCP) and Human Subject Protection (HSP) issues arising in human research trials regulated by FDA.
Clinical Study Initiation
The following is needed by a sponsor to initiate a clinical study:
- IRB
- Documentation of the clinical investigator’s credentials
- Financial disclosure (grant-sponsor)
- GCP assurance statements
- Verification of study protocol training
Clinical Study Reporting
The investigator must provide clinical study progress reports at specified intervals during the study.
Clinical Study Design
A clinical study is any research study that involves one or more human subjects testing experimental new drugs, devices, or biologics (or control). It is the investigator's job to design the clinical study protocol. There are two main types of clinical trials; clinical (interventional) studies, and observational studies. For certain medical devices, accuracy studies may also be appropriate. Within the clinical study type, there are several subtypes, which may include placebo-control, double-blind studies, and randomization controls.
Test Your Knowledge!
Beth is a 46-year-old post-menopausal mentally disabled woman with LCIS, meaning, she is predisposed to develop breast cancer later in life. Her caregivers with power of attorney for health care decisions, bring her to the clinic for enrollment in a clinical trial, which is a randomized trial of tamoxifen v. raloxifene for the prevention of breast cancer in high-risk women. She fulfills all entry requirements but cannot consent due to her mental disability. The IRB is considering, is it ethical to permit Beth’s power of attorney to enroll her in this study? *Remember, we are focusing on ethics here – not the law*.
- What is an IRB? What is their function?
- Using what you’ve learned in this chapter on GCPs and the ethics resources below, argue for OR against the IRB ruling to grant Beth’s power of attorney permission to enroll her in the study.
Clinical Trials
During a clinical trial, participants may receive a particular intervention, an investigational new drug, device or biologic, or even psychological treatment, for example, diet or quit smoking. These interventions may be a comparison of a current drug to a new investigational drug, a placebo with no active ingredient to an existing drug, to name a few examples. The trial may also be randomized, placebocontrol, and blinded to reduce study bias.
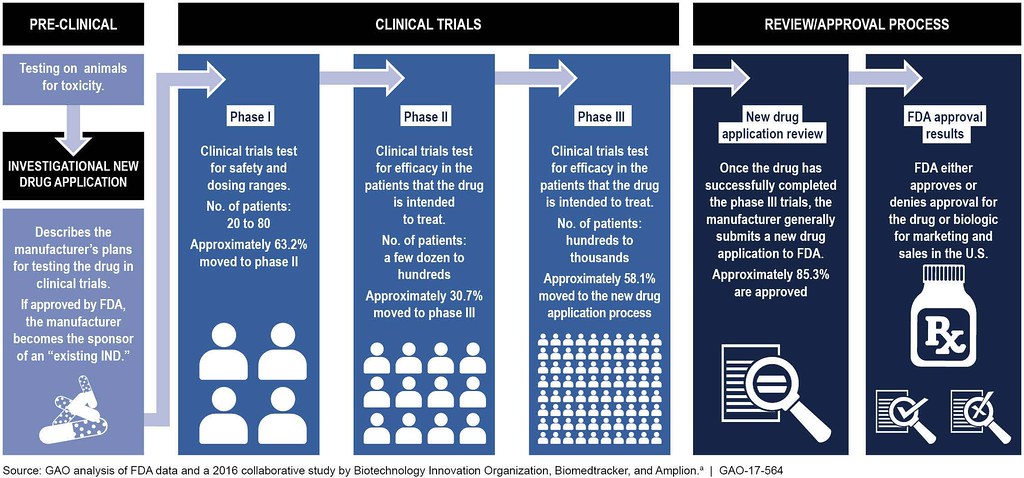
The Four Phases of Clinical Trials
- In Preclinical Studies, the drug is tested on animals for safety and efficacy.
- Phase I clinical trials primarily test for the safety of the proposed drug in healthy humans. During Phase I trials, the drug is administered to 20-80 healthy volunteers who will report any unexpected side effects and help establish the dosage levels that can be tolerated. In addition to evaluating the safety of the drug, its metabolic and pharmacologic properties in healthy humans are determined. If a drug meets the safety requirements at this phase and appears to have the desired impact of treatment, then it enters Phase II clinical trials.
- Phase II trials are performed on a small number of patients to determine the drug's efficacy. Between 100 and 300 patients that the drug is intended to treat are given various dosages. Clinical trial participants are carefully monitored for side effects as well as the consequences of the drug treatment. If there no detrimental side effects, and the drug has a positive effect, it goes to phase III.
- Phase III trials involve between 1,000 and 3,000 patients in double-blind studies usually conducted across several hospital sites. For most new drugs, these tests will last three or more years to establish the drug's benefits, recommended dosage, and long-term safety. Additional data on drug-drug interactions and risks versus benefits are collected.
- Phase IV trials are performed after the drug has been approved by the FDA. This post-market surveillance help gathers additional information on drug safety and efficacy by the general population.
Medical Device Clinical Trials
Not all medical devices undergo clinical trial testing. Minimal risk devices such as bandages (Class I) do not require clinical trials, where Class II devices of intermediate risk may depend on the device. Class III devices have a substantive risk and therefore undergo clinical trials. Another difference between medical device clinical trials is what is tested. For drugs, a dosage is tested; however, in devices the prototype is. Since there is an array of types of medical devices, we will further explore this in a later chapter focusing on medical devices.
The NIH has a public-accessible registry of all clinical trials currently underway and includes the results, appropriately named ClinicalTrials.gov.
Explore!
Go to ClinicalTrials.gov and “search for a study” of something that interests you. For example, if you have been following the latest Ebola or Zika virus outbreak, you may wonder about the status of any current vaccine study. Write a 5-sentence summary of the study you selected.