
SS1_2019_Lecture_10
- Page ID
- 23938
Genomes as organismal blueprints
A genome, not to be confused with a gnome, is an organism's complete collection of heritable information stored in DNA. Differences in information content help to explain the diversity of life we see all around us. Changes to the information encoded in the genome are the primary drivers of the phenotypic diversity we see (and some we can't) around us that are filtered by natural selection, and they are thus the drivers of evolution. This leads to questions. If every cell in a multicellular organism contains the same sequence of DNA, how can there be different cell types (e.g., how can a cell in a liver be so different from a cell in the brain if they both carry the same DNA)? How do we read the information? How do we interpret what we read? How do we understand how all of the "parts" we identify in the genome functionally interrelate? How is all of this related to the expression of traits? How do changes in the genome lead to changes in traits?
Determining a genome sequence
The information encoded in genomes provides important data for understanding life, its functions, its diversity, and its evolution. Therefore, it stands to reason that a reasonable place to begin studies in biology would be to read the information content encoded in the genome(s) in question. A good starting point is to determine the sequence of nucleotides (A, G, C, T) and their organization into one or more independently replicating units of DNA (e.g., think chromosomes and/or plasmids ). For 30+ years after the discovery that DNA is the hereditary material, this was a daunting proposition. In the late 1980s, however, the advent of semi-automated tools for DNA sequencing were pioneered, and this began a revolution that has dramatically changed how we approach the study of life. Twenty years later, in the mid-2000s, we entered a period of accelerated technological progress in which advances in materials sciences (particularly, advances in our ability to make things on a very small scale), optics, electrical and computer engineering, bioengineering, and computer sciences have all converged to bring us dramatic increases in our capacity to sequence DNA and correspondingly dramatic decreases in the cost of numerous advances in our ability to sequence DNA. A famous example to illustrate this point is to compare the changes in cost to sequence the human genome. The first draft of the human genome took nearly 15 years and $3 billion dollars to complete. Today, 10's of human genomes can be sequenced in a single day on a single instrument at a cost of less than $1000 each (the cost and time continue to decrease). Today, companies like Illumina, Pacific Biosciences, Oxford Nanopore, and others offer competing technologies that are driving down the cost and increasing the volume, quality, speed, and portability of DNA sequencing.
One of the very exciting elements of the DNA sequencing revolution is that it has required and continues to require contributions from biologists, chemists, materials scientists, electrical engineers, mechanical engineers, computer scientists and programmers, mathematicians and statisticians, product developers, and many other technical experts. The potential applications and implications of unlocking barriers to DNA sequencing have also engaged investors, business people, product developers, entrepreneurs, ethicists, policy makers, and many others to pursue new opportunities and to think about how to best and most responsibly use this growing technology.
The technological advances in genome sequencing have resulted in a virtual flood of complete genome sequences being determined and deposited into publicly available databases. You can find many of them at the National Center for Biotechnology Information. The number of available , completely sequenced genomes numbers in the tens of thousands—over 2,000 eukaryotic genomes, over 600 archaeal genomes, and nearly 12,000 bacterial genomes at the time of this writing. Tens of thousands of more genome sequencing projects are in progress. With this many genome sequences available—or soon to be available—we can start asking many questions about what we see in these genomes. What patterns are common to all genomes? How many genes are encoded in genomes? How are these organized? How many different types of features can we find? What do the features that we find do? How different are the genomes from one another? Is there evidence that can tell us how genomes evolve? Let's briefly examine a few of these questions.
Diversity of genomes
Diversity of sizes, number of genes, and chromosomes
Let's start by examining the range of genome sizes. In the table below, we see a sampling of genomes from the database. We can see that the genomes of free living organisms range tremendously in size. The smallest known genome is encoded in 580,000 base pairs while the largest is 150 billion base pairs—for reference, recall that the human genome is 3.2 billion base pairs. That's a huge range of sizes. Similar disparities in the number of genes also exist.
Table 1. This table shows some genome data for various organisms. 2n = diploid number. Attribution: Marc T. Facciotti (own work—reproduced from http://book.bionumbers.org/how-big-are-genomes/)
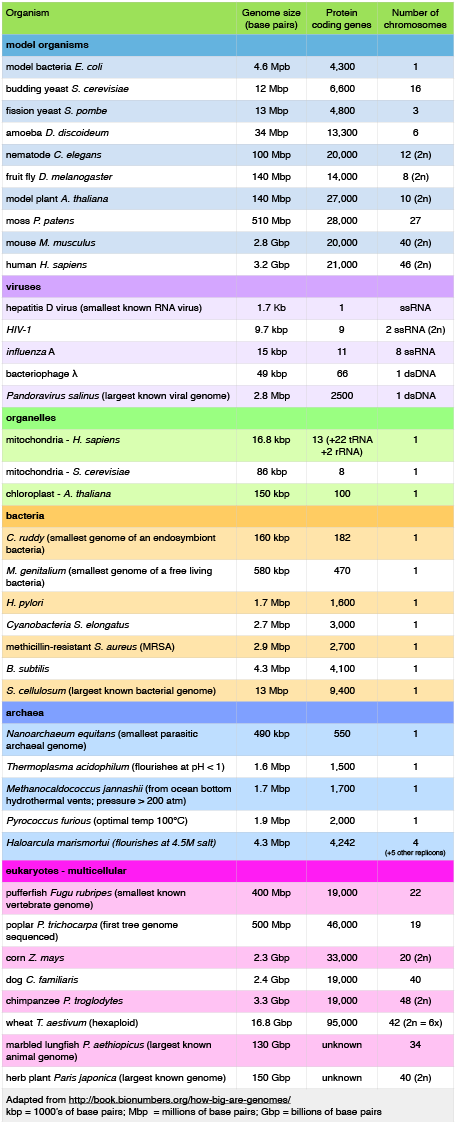
Examining Table 1 also reveals that some organisms carry with them more than one chromosome. Some genomes are also polyploid, meaning that they maintain multiple copies of similar but not identical (homologous) copies of each chromosome. A diploid organism carries in its genome two homologous copies (usually one from Mom and one from Dad) of each chromosome. Humans are diploid. Our somatic cells carry 2 homologous copies of 23 chromosomes. We received 23 copies of individual chromosomes from our mother and 23 copies from our father, for a total of 46. Some plants have higher ploidy. For example, a plant with four homologous copies of each chromosome is termed tetraploid. An organism with a single copy of each chromosome is termed haploid.
Structure of genomes
Table 1 also provides clues to other points of interest. For instance, if we compare the pufferfish genome to the chimpanzee genome, we note that they encode roughly the same number of genes (19,000), but they do so on dramatically differently sized genomes—400 million base pairs versus 3.3 billion base pairs, respectively. That implies that the pufferfish genome must have much less space between its genes than what might be expected to be found in the chimpanzee genome. Indeed, this is the case, and the difference in gene density is not unique to these two genomes. If we look at Figure 1, which attempts to represent a 50-kb part of the human genome, we notice that in addition to the protein-coding regions (indicated in red and pink) that many other so-called "features" can be read from the genome. Many of these elements contain highly repetitive sequences.
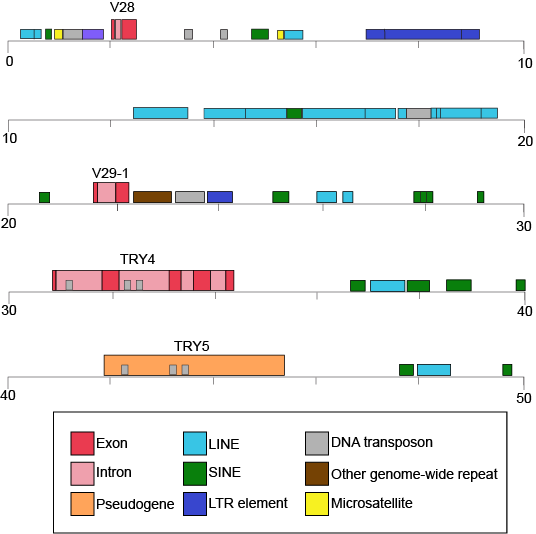
Figure 1. This figure shows a 50-kb segment of the human β T-cell receptor locus on chromosome 7. This figure depicts a small region of the human genome and the types of "features" that can be read and decoded in the genome, including, but also in addition to, protein-coding sequences. Red and pink correspond to regions that encode proteins. Other colors represent different types of genomic elements. Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
If we now look at what fraction of the whole human genome each of these types of elements makes up (see Figure 2), we see that protein-coding genes only make up 48 million of the 3.2 billion bases of the haploid genome.
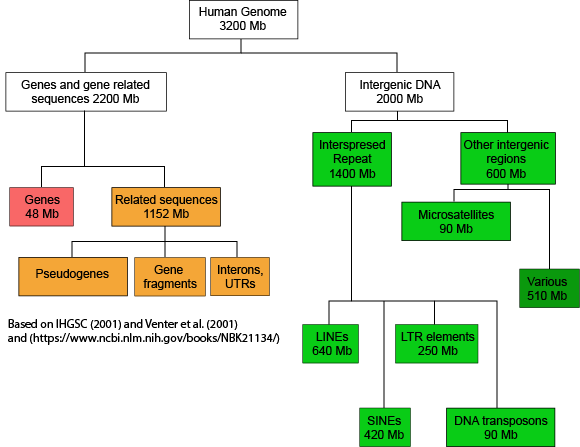
Figure 2. This graph depicts how the many base pairs of DNA in the human haploid genome are distributed between various identifiable features. Note that only a small fraction of the genome is associated directly with protein-coding regions. Attribution: Marc T. Facciotti (own work—reproduced from sources noted in figure)
When we examine the frequency of repeat regions versus protein-coding regions in different species, we note large differences in protein-coding versus non-coding regions.
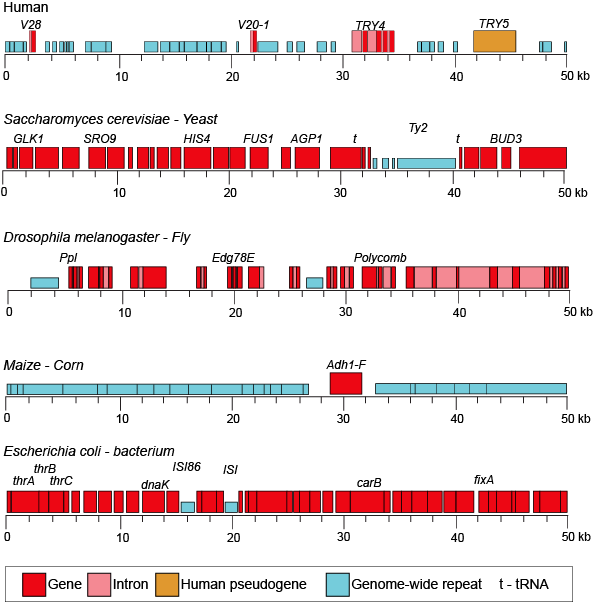
Figure 3. This figure shows 50-kb segments of different genomes, illustrating the highly variable frequency of repeat versus protein-coding elements in different species.
Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
Suggested discussion
Propose a hypothesis for why you think some genomes might have more or fewer noncoding sequences.
Dynamics of genome structure
Genomes change over time, and numerous different types of events can change their sequence.
1. Mutations are either accumulated during DNA replication or through environmental exposure to chemical mutagens or radiation. These changes typically occur at the level of single nucleotides.
2. Genome rearrangements describe a class of large-scale changes that can occur, and they include the following: (a) deletions—where segments of the chromosome are lost; (b) duplication—where regions of the chromosome are inadvertently duplicated; (c) insertions—the insertion of genetic material (note that sometimes this is acquired from viruses or the environment, and deletion/insertion pairs may happen across chromosomes); (d) inversions—where regions of the genome are flipped within the same chromosome; and (e) translocations—where segments of the chromosome are translocated (moved elsewhere in the chromosome).
These changes happen at different rates, and some are facilitated by the activity of enzyme catalysts (e.g., transposases).
The study of genomes
Comparative genomics
One of the most common things to do with a collection of genome sequences is to compare the sequences of multiple genomes to one another. In general terms, these types of activities fall under the umbrella of a field called comparative genomics.
Comparing the genomes of people who suffer from an inheritable disease to the genomes of people who are not afflicted can help us to uncover the genetic basis for the malady. Comparing the gene content, order, and sequence of related microbes can help us find the genetic basis of why some microbes cause disease while their close cousins are virtually harmless. We can compare genomes to understand how a new species may have evolved. There are many possible analyses! The basis of these analyses is similar: look for differences across multiple genomes and try to associate those differences with different traits or behaviors in those organisms.
Lastly, some people are comparing genome sequences to try to understand the evolutionary history of the organisms. Typically, these types of comparisons result in a graph known as a phylogenetic tree, which is a graphical model of the evolutionary relationship between the various species being compared. This field, not surprisingly, is called phylogenomics.
Metagenomics: who is living somewhere and what are they doing?
In addition to studying the genomes of individual species, the increasingly powerful DNA-sequencing technologies are making it possible to simultaneously sequence the genomes of environmental samples that are inhabited by many different species. This field is called metagenomics. These studies are typically focused on trying to understand what microbial species inhabit different environments. There is great interest in using DNA sequencing to study the populations of microbes in the gut and to watch how the population changes in response to different diets, to see if there is any association between the abundance of different microbes and various diseases, or to look for the presence of pathogens. People are using DNA sequencing of environmental metagenomic samples to explore which microbes inhabit different environments on Earth (from the deep sea, to soil, to air, to hypersaline ponds, to cat feces, to some of the common surfaces we touch every day).
In addition to discovering "who lives where," the sequencing of microbial populations in different environments can also reveal what protein-coding genes are present in an environment. This can give investigators clues into what metabolic activities might be occurring in that environment. In addition to providing important information about what kind of chemistry might be happening in a specific environment, the catalog of genes that is accumulated can also serve as an important resource for the discovery of novel enzymes for applications in biotechnology.
The DNA Double Helix and Its Replication
The scope of the problem
In this module we discuss the replication of DNA—one of the key requirements for a living system to regenerate and create the next generation. Let us first briefly consider the scope of the problem by way of a literary analogy.
The human genome consists of roughly 6.5 billion base pairs of DNA if one considers the full diploid genome (i.e., if you count the DNA inherited from both parents). Six point five billion looks like this: 6,500,000,000. That's a large number. To get a better idea of what that number means, imagine that our DNA is a set of written instructions for constructing one of us. By analogy we can then compare it to another written document. For this example we begin by considering Tolstoy's War and Peace, a novel many people are familiar with for its voluminous nature. Data from Wikipedia estimates that War and Peace contains about 560,000 words. A second written work many are familiar with are the seven volumes of J.K. Rowling's Harry Potter. This work checks in at ~1,080,000 words (Referenced Statistics on Wikipedia). If we assume that the length of the average English word is five characters, the two literary works are 2.8 million and 5.4 million characters in length, respectively. Therefore, even all seven volumes of "Harry Potter" have over 1000x fewer characters than our own genomes. The number of characters in these novels are, however, much closer to the number of nucleotides in a typical bacterial genome.
Now imagine for a moment developing a machine or mechanical process (not an electronic process) that is responsible for reading and copying these books. Or imagine yourself copying these texts. How fast could you do it? How many mistakes are you likely to make? Do you expect there to be a trade-off between the speed at which you can copy and the accuracy? What type of resources does this process need? How much energy is required? Now imagine copying something 1000x larger! Oh, and just for good measure, your imaginary mechanical device needs to do its work on text that is ~25Å wide (i.e., 0.0000000025 meters wide). By comparison, a typical ten point font is ~0.00025 meters wide, about 100,000x larger than with width of a DNA base pair.
With that in mind, it is worth noting that a human cell can take about 24 hours to divide (DNA replication must therefore be a little faster). A healthy E. coli cell may take only 20 minutes to divide (including replicating its ~4.5 million base pair genome). Both the human and bacterium do this while typically making few enough mistakes that the subsequent generation remains viable and recognizable. That should seem rather amazing! Now consider that the human body is estimated to consist of ~10 trillion cells (10,000,000,000,000) and that it may have between two and ten times that number of microbial residents and that's a lot of cell division to consider.
Design challenge
If the cell is to replicate—its ultimate goal—a copy of the DNA must be created. So one clear problem statement/question is "how can the cell effectively copy its DNA?" Given the analogy above, here are some relevant sub-questions: What are the chemical and physical properties that enable DNA to be copied? With what fidelity must the DNA be copied? What speed must it be copied at? Where does the energy come from for this task and how much is necessary? Where do the "raw materials" come from? How do the molecular machines involved in this process couple the assembly of raw materials and the energy required to build a new DNA molecule together? The list could, of course, go on.
In the following discussion and in lecture we will be interested in starting to examine how the process of DNA replication is accomplished while keeping in mind some of the driving questions. As you go through the reading and lecture materials, try to constantly be aware of these and other questions associated with this process. Use these questions as guideposts for organizing your thoughts and try to find matches between the "facts" that you think you might be expected to know and the driving questions.
The DNA double helix

To build some extra context we also need a little bit of empirically determined knowledge. Perhaps one of the best known and popular features of the hereditary form of the DNA molecule is that it has a double helical tertiary structure. The appreciation of this dates to the the 1950s. The story of this discovery has been widely recounted—and the details are beyond the scope of this text. Briefly, Francis Crick and James Watson are credited with determining the structure of DNA. Rosalind Franklin is now also widely credited with generating critical X-ray diffraction data that enabled Watson and Crick to piece together the puzzle of the DNA molecule.
Models of the structure of DNA revealed the molecule is made up of two strands of covalently linked nucleotides that are twisted around each other to form a right-handed helix. In each strand, nucleotides are covalently joined to two other nucleotides (except at the very ends of a linear strand) via phosphodiester bonds that link the sugars via the 5' and 3' hydroxyl groups (panel b in Figure 1). Recall that the labels 5' and 3' refer to the carbons on the sugar molecule. These sugar and phosphate chains form a contiguous set of covalent links that are often referred to as the "backbone" of the structure. In a linear molecule, each strand has two free ends. One is termed the 5' end because the unlinked functional group that is typically involved in joining nucleotides is the phosphate linked to the 5' carbon. The other end of the strand is called the 3' end because the unlinked functional group that is typically involved in joining nucleotides is the hydroxyl group linked to the 3' carbon of the sugar. Since the two ends of the strand are not symmetrical, this makes it easy to designate or describe a direction one the strand—one can, for instance, say that they are reading from the 5' end to 3' end to indicate that they are "walking" along the strand starting at the 5' end and and moving towards the 3' end. This direction (5' to 3') is, by the way, the convention used by most biologists. One can read in the opposite direction (3' to 5') provided this is made explicit. The two strands of covalently linked nucleotides are found to be anti-parallel to one another in the double-helix; that is, the orientation/direction of one strand is opposite to that of the other strand (panel b in Figure 1). The backbone is structurally on the "outside" of the double helix, creating a band of negative charges on the surface. By contrast, the nitrogenous bases of each of the antiparallel strands stack on the inside of the structure and oppose one another in a way that allows hydrogen bonds between unique purine/pyrimidine pairs (A pairing with T and G pairing with C) to form. These specific pairings are said to be complementary to on another and thus the opposite strands of a double helix are often referred to one another as complementary strands.
Complementary strands carry redundant information. Because of the strict chemical pairing, if you know the sequence of one strand you obligatorily know the strand of its complement. Take for example the sequence 5′- C A T A T G G G A T G - 3′. Note how the sequence is annotated with the orientation (indicated by 5' and 3' labels). The complement of this sequence—written according to the 5' to 3' convention is: 5′- C A T C C C A T A T G - 3′. If you aren't convinced, write these two sequences out across from one another in your notes, making sure to write them as antiparallel strands. Note that the twisting of the two complementary strands around each other results in the formation of structural features called the major and minor grooves that will become more important when we discuss the binding of proteins to DNA (panel c in Figure 1).
Most of the BIS2A instructors will expect you to recognize key structural features depicted in the figure below and that you will be able to create a basic figure of the structure of DNA yourself.
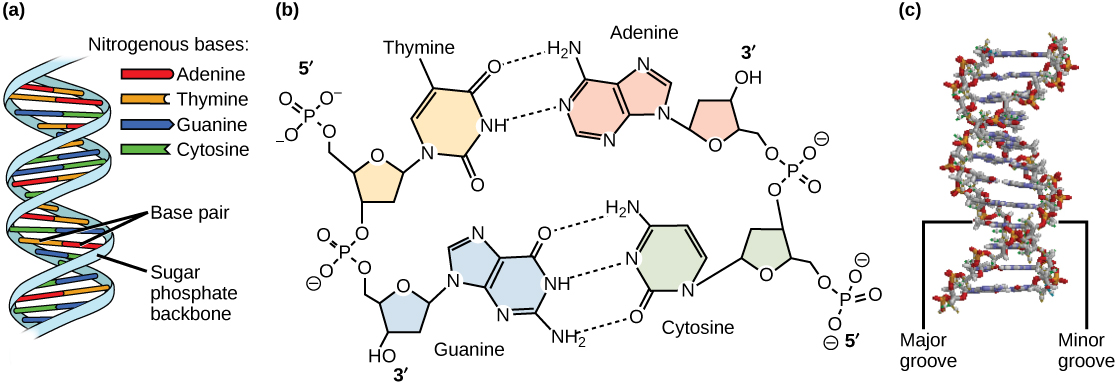
Figure 1. DNA has (a) a double helix structure and (b) phosphodiester bonds. The (c) major and minor grooves are binding sites for DNA binding proteins during processes such as transcription (the creation of RNA from a DNA template) and replication.
At around the same time, three hypotheses for the modes of DNA replication were being considered. The models for replication were known as: the conservative model, the semi-conservative model, and the dispersive model.
1. Conservative: The conservative model of replication postulated that each whole double-stranded molecule could act as a template for the synthesis of a completely new double-stranded molecule. That is, if one were to put a chemical tag on the template DNA molecule after replication none of that tag would be found on the new copy.
2. Semi-conservative: This hypothesis stipulated that each individual strand of a DNA molecule could serve as a template for a new strand to which it would now associate with. in this case, if a chemical label were placed on a double-stranded DNA molecule, one strand on each of the copies would retain the label.
3. Dispersive: This model proposed that a copied double helix would be a piecewise combination of continuous segments of "old" and "new" strands. If a chemical label were placed on a DNA molecule that were copied using a dispersive mechanism, one would find discrete segments of the resulting copy that were labeled on both strands separated by completely unlabeled parts.
Meselson and Stahl resolved the issue in 1958 when they reported results of a now famous experiment (describe on Wikipedia) which showed that DNA replication is semi-conservative (Figure 2), where each strand is used as a template for the creation of the new strand. To learn more about this experiment watch The Meselson-Stahl Experiment.
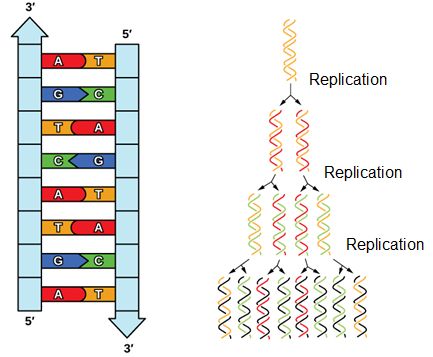
Figure 2. DNA has an antiparallel double helix structure, the nucleotide bases are hydrogen bonded together and each strand complements the other. DNA is replicated in a semi-conservative manner, each strand is used as the template for the newly made strand.
DNA replication
Having established some basic structural features and the need for a semi-conservative mechanism, it is important to begin understanding some of what is known about the process and to think about what questions one might want to answer if they are to better understand what is going on.
Since DNA replication is a process, we can invoke the energy story rubric to think about it. Recall that the energy story rubric is there to help us think systematically about processes (how things go from A to B). In this case the process in question is the act of starting with one double-stranded DNA molecule and ending up with two double-stranded molecules. So, we will ask a variety of questions: What does the system look like at the beginning (matter and energy) of replication? How are matter and energy transferred in the system and what catalyzes the transfers? What does the system look like at the end of the process? We can also ask questions regarding specific events that MUST happen during the process. For instance, since DNA is a long molecule and it is sometimes circular, we can ask basic questions like, where does the process of replication start? Where does it end? We can also ask practical questions about the process like, what happens when a double-stranded structure is unwound?
We consider some of these key questions in the text and in class and encourage you to do the same.
Requirements for DNA replication
Let's start by listing some basic functional requirements for DNA replication that we can infer just by thinking about the process that must happen and/or be required for the replication to happen. So, what do we need?
• We know that DNA is composed of nucleotides. If we are going to create a new strand, we will need a source of nucleotides.
• We can infer that building a new strand of DNA will require an energy source—we should try to find this.
• We can infer that that there must be a process for finding a place to start replication.
• We can infer that there will be one or more enzymes that help catalyze the process of replication.
• We can also infer that since this is a biochemical process, that some mistakes will be made.
Nucleotide structure review
Recall some basic structural features of the nucleotide building blocks of DNA. The nucleotides start off as nucleotide triphosphates. A nucleotide are composed of a nitrogenous base, deoxyribose (five-carbon sugar), and a phosphate group. The nucleotide is named according to its nitrogenous base, purines such as adenine (A) and guanine (G), or pyrimidines such as cytosine (C) and thymine (T). Recall the structures below. Note that the nucleotide Adenosine triphosphate (ATP) is a precursor of the deoxyribonucleotide (dATP) which is incorporated into DNA.

Initiation of replication
Where along the DNA does the replication machinery start DNA replication?
With millions, if not billions, of nucleotides to copy how does the DNA polymerase know where to start? Not surprisingly, this process turns out not to be random. There are specific nucleotide sequences called origins of replication along the length of the DNA at which replication begins. Once this site is identified, however, there is a problem. The DNA double helix is held together by base stacking interactions and hydrogen bonds. If each strand is to be read and copied individually, there must be some mechanism responsible for helping to dissociate the two strands from one another. Energetically, this is an endergonic process. Where does the energy come from and how is this reaction catalyzed? Basic reasoning should, at this point, lead to the hypothesis that a protein catalyst is likely involved, and this enzyme either creates new bonds that are energetically more favorable (exergonic) than the bonds it breaks AND/OR it is able to couple the use of an external energy source to help dissociate the strands.
It turns out that the details of this process and the proteins involved differ depending on the specific organism in question, and many of the molecular level details are yet to be completely understood. There are, however, some common features in the replication of eukaryotes, bacteria, and archaea, and one of these features is that multiple different types of proteins are involved in replicating DNA. First, proteins generally called "initiators" have the capacity to bind DNA at or very near origins of replication. The interaction of the initiator proteins with the DNA helps to destabilize the double helix and also help to recruit other proteins, including an enzyme called a DNA helicase to the DNA. In this case the energy required to destabilize the DNA double helix seems to come from the formation of new associations between DNA and the initiator proteins and the proteins themselves. The DNA helicase is a multi-subunit protein that is important in the process of replication because it couples the exergonic hydrolysis of ATP to the unwinding of the DNA double helix. Additional proteins must be recruited to the initiation complex (the collection of proteins involved in initiating transcription). These include, but are not limited to, additional enzymes called primase and DNA polymerase. While the initiators are lost soon after the initiation of replication, the rest of the proteins work in concert to execute the process of DNA replication. This complex of enzymes function at Y-shaped structures in the DNA called replication forks (Figure 4). For any replication event two replication forks may be formed at each origin of replication, extending in both directions. Multiple origins of replication can be found on eukaryotic chromosomes and some archaea, while the the genome of the bacterium, E. coli, seems to encode one origin of replication.
Note: possible discussion
Why would different organisms have different numbers of replication origins? What could the benefit be to having more than one? Is there a drawback to having more than one?
Note: possible discussion
Given what needs to happen at origins of replication, can you use logic to infer and propose for discussion some potential features that distinguish replication origins from other segments of DNA?
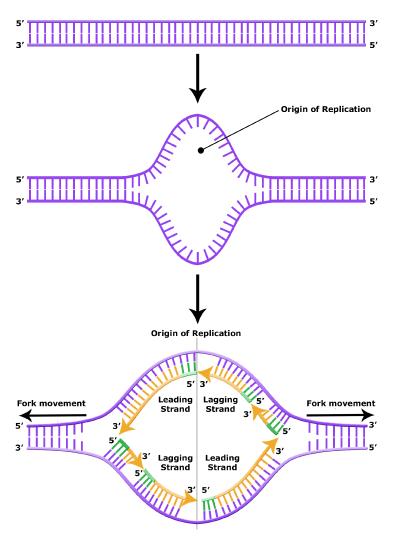
Elongation of replication
The melting open of the DNA double helix and the assembling the DNA replication complex is just the first step in the process of replication. Now the process of creating a new strand actually needs to get started. Here additional challenges are encountered. The first obvious issue is that of determining which of the two strands should be copied at any replication fork (i.e., Which strand will serve as a template for semi-conservative synthesis? Are both strands equally viable alternatives?). There is also the problem of actually getting the process of the new strand synthesis started. Can the DNA polymerase initiate the new strand on its own? The answer to the latter question, and some of the rationale and consequences, will be discussed later. The key idea to note at this point is that it has been experimentally determined that DNA polymerase can NOT initiate strand synthesis on its own. Rather, DNA polymerase requires a short stretch of double-stranded structure followed by single-stranded template. The creation of a short oligonucleotide is carried out by the enzyme primase. This protein creates a short polymer of RNA (not DNA) called a primer (these are depicted by short green lines in the figures above and below) that can be used by DNA polymerase to nucleate a new growing strand.
During the process of strand elongation, the DNA polymerase polymerizes a new covalently-linked strand of DNA nucleotides (in bacteria this specific enzyme may be called DNA polymerase III; in eukaryotes, polymerase nomenclature is more complex and the roles of several polymerase proteins are not completely understood). It turns out that one of the strands is favored exclusively over the other to serve as a template. DNA polymerase will "read" the template strand from 3' to 5' and synthesize a new strand in the 5' to 3' direction. Hypotheses to explain this universal observation usually center around the energetics associated with the addition of a new nucleotide and arguments associated with DNA repair that we will describe shortly. Let us, therefore, briefly consider the reaction involving the addition of a single nucleotide. The primer provides an important 3' hydroxyl on which to begin synthesis. The next deoxyribonucleotide triphosphate enters the binding site of the DNA polymerase and, as shown in Figure 5 below, is oriented by the polymerase such that a hydrolysis of the 5' triphosphate can occur, releasing pyrophosphate and coupling this exergonic reaction to the synthesis of a phosphodiester bond between the 5' phosphate of the incoming nucleotide and the 3' hydroxyl group of the primer. This process can be repeated until deoxyribonucleotide triphosphates run out or the replication complex falls off of the DNA. In effect, DNA polymerase adds the phosphate group (5') from the incoming nucleotide to the existing hydroxyl group (3') of the previously added nucleotide.
Correct base pairing, or selection of correct nucleotide to add at each step, is accomplished by structural constraints felt by the DNA polymerase and the energetically favorable hydrogen bonds formed between complementary nucleotides. The process is energetically driven by the hydrolysis of the incoming 5' triphosphate and the energetically favorable interactions formed by the inter-nucleotide interactions in the growing double helix (base stacking and complementary base pairing hydrogen bonds). Note that the energetics of nucleotide addition do not technically preclude a strand growing in the 3' to 5' direction, the key difference in this scheme is that the energy "source" for synthesis would need to come from a nucleotide already incorporated into the growing strand rather than the new incoming nucleotide (which this might be an important selective disadvantage is discussed briefly). After elongation has started a different DNA polymerase (in bacteria this is usually called DNA Polymerase I) comes in to remove the RNA primer and to synthesize the remaining bit of missing DNA.
As will be discussed in more detail in class, the movement of the replication fork induces winding of the DNA in both directions of replication. Another ATP consuming enzyme called topoisomerase helps to relieve this stress.
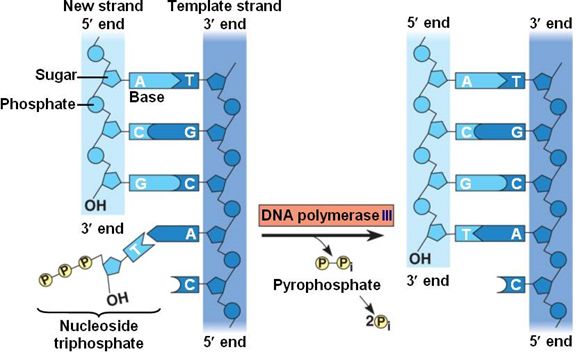
Figure 5. DNA polymerase catalyzes the addition of the 5' phosphate group from an incoming nucleotide to the 3' hydroxyl group of the previous nucleotide. This process creates a phosphodiester bond between the nucleotides while hydrolyzing the phosphoanhydride bond in the nucleotide.
Source: http://bio1151.nicerweb.com/Locked/m...h16/elong.html
Note: possible discussion
Create an energy story for the addition of a nucleotide onto a polymer as shown in the figure above. This will be an explicit learning goal from some of your BIS2A instructors.
Leading and lagging strand
The discussion above about strand elongation describes the process of new strand synthesis if that strand happens to be synthesized in the same direction as the replication fork is or appears to be moving along the DNA. This strand can be synthesized continuously and is called the leading strand. However, both strands of the original DNA double helix must be copied. Since the DNA polymerase can only synthesize DNA in a 5' to 3' direction, the polymerization of the strand opposite of the leading strand must occur in the opposite direction that helicase, or front of the replication fork, is traveling. This strand is called the lagging strand, and due to geometric constraints, must be synthesized through a series of RNA priming and DNA synthesis events into short segments called Okazaki fragments. As noted, the initiation of synthesis of each Okazaki fragment requires a primase to synthesize an RNA primer, and each of these RNA primers must be ultimately removed and replaced with DNA nucleotides by a different DNA polymerase. The covalent bonds between each of the Okazaki fragment can not be made by the DNA polymerase and must therefore be formed by yet another enzyme called DNA ligase. The geometry of lagging strand synthesis is difficult to visualize and will be covered in class.
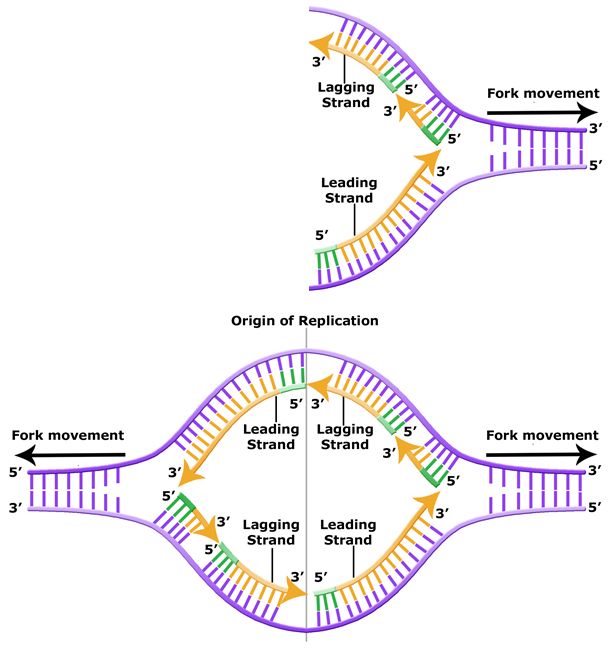
Figure 6. The lagging strand is created in multiple segments. A replication fork shows the leading and lagging strand. A replication bubble shows the leading and lagging strands.
BIS2A Team original image
Termination of replication
Telomeres and telomerase
The ends of replication in circular bacterial chromosomes poses few practical problems. However, the ends of linear eukaryotic chromosomes pose a specific problem for DNA replication. Because DNA polymerase can add nucleotides in only one direction (5' to 3'), the leading strand allows for continuous synthesis until the end of the chromosome is reached; however, as the replication complex arrives at the end of the lagging strand there is no place for the primase to "land" and synthesize an RNA primer so that the synthesis of the missing lagging strand DNA fragment at the end of the chromosome can be initiated by the DNA polymerase. Without some mechanism to help fill this gap, this chromosomal end will remain unpaired and will be lost to nucleases. Over time, and several rounds of replication, this would result in the ends of linear chromosomes getting progressively shorter, ultimately compromising the ability of the organism to survive. These ends of the linear chromosomes are known as telomeres, and nearly all eukaryotic species have evolved repetitive sequences that do not code for a specific gene. As a consequence, these "non-coding" telomeres act as replication buffers and are shortened with each round of DNA replication instead of critical genes. For example, in humans, a six base-pair sequence, TTAGGG, is repeated 100 to 1000 times at the end of most chromosomes. In addition to acting as a potential buffer, the discovery of the enzyme telomerase helped in the understanding of how chromosome ends are maintained. Telomerase is an enzyme composed of protein and RNA. Telomerase attaches to the end of the chromosome by complementary base pairing between the RNA component of telomerase and the DNA template. The RNA is used as a complementary strand for the short elongation of its complement. This process can be repeated numerous times. Once the lagging strand template is sufficiently elongated by telomerase, primase will create a primer followed by DNA polymerase which can now add nucleotides that are complementary to the ends of the chromosomes. Thus, the ends of the chromosomes are replicated.

Figure 7. The ends of linear chromosomes are maintained by the action of the telomerase enzyme.
Telomerase is not active in adult somatic cells. Adult somatic cells that undergo cell division continue to have their telomeres shortened. This essentially means that telomere shortening is associated with aging. In 2010, scientists found that telomerase can reverse some age-related conditions in mice, and this may have potential in regenerative medicine.1 Telomerase-deficient mice were used in these studies; these mice have tissue atrophy, stem-cell depletion, organ system failure, and impaired tissue injury responses. Telomerase reactivation in these mice caused extension of telomeres, reduced DNA damage, reversed neurodegeneration, and improved functioning of the testes, spleen, and intestines. Thus, telomere reactivation may have potential for treating age-related diseases in humans.
Differences in DNA replication rates between bacteria and eukaryotes
DNA replication has been extremely well studied in bacteria, primarily because of the small size of the genome and large number of variants available. E. coli has 4.6 million base pairs in a single circular chromosome, and all of it gets replicated in approximately 42 minutes, starting from a single origin of replication and proceeding around the chromosome in both directions. This means that approximately 1000 nucleotides are added per second. The process is much more rapid than in eukaryotes. Table 1 summarizes the differences between bacterial and eukaryotic replications.
Table 1. Differences between bacterial and eukaryotic replication
| Differences between prokaryotic and eukaryotic replication | ||
|---|---|---|
| Property | Bacteria | Eukaryotes |
| Origin of replication | Single | Multiple |
| Rate of polymerization per polymerase | 1000 nucleotides/s | 50 to 100 nucleotides/s |
| Chromosome structure | Circular | Linear |
| Telomerase | Not present | Present |
Link to external resource
Click through a tutorial on DNA replication.