
B1. Amino Acid Analysis and Chemical Sequencing
- Page ID
- 4717

As described in the Introduction to Proteins, we can understand proteins structure at varying level of complexity.
Figure: Protein Analysis from low to high resolution.
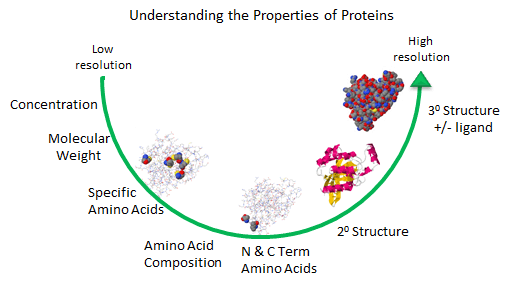
In the last chapter section, we learned about the charge and chemical reactivity properties of isolated amino acids and amino acids in proteins. The analysis of a whole protein is complicated since each different amino acid might be represented many times in the sequence. Each protein has an N-terminal and C-terminal amino acid and secondary structure. Some proteins exists biologically as multisubunit proteins, which adds to the complexity of the analyses since now the proteins would have multiple N- and C-terminal ends. In addition, isolated proteins might have chemically modifications (post-translational) which add to the functionalities of the proteins but also add to the complexities of the analyses. To illustrate some of these issues, view the structure of the RhoA program below.
 Updated RhoA - a cytoplasmic protein - The complexity of protein analysis Jmol14 (Java) | JSMol (HTML5)
Updated RhoA - a cytoplasmic protein - The complexity of protein analysis Jmol14 (Java) | JSMol (HTML5)
Amino Acid Composition
At a low level of resolution, we can determine the amino acid composition of the protein by hydrolyzing the protein in 6 N HCl, 100oC, under vacuum for various time intervals. After removing the HCl, the hydrolysate is applied to an ion-exchange or hydrophobic interaction column, and the amino acids eluted and quantitated with respect to known standards. A non naturally- occurring amino acid like norleucine is added in known amounts as an internal standard to monitor quantitative recovery during the reactions. The separated amino acids are often derivitized with ninhydrin or phenylisothiocyantate to facilitate their detection. The reaction is usually allowed to procedure for 24, 36, and 48 hours, since amino acids with OH (like ser) are destroyed. A time course allows the concentration of Ser at time t=0 to be extrapolated. Trp is also destroyed during the process. In addition, the amide links in the side chains of Gln and Asn are hydrolyzed to form Glu and Asp, respectively.
- AA Analysis: Iowa State University Protein Facility
N- and C-Terminal Amino Acid Analysis
The amino acid composition does not give the sequence of the protein. The N-terminus of the protein can be determined by reacting the protein with fluorodinitrobenzene (FDNB) or dansyl chloride, which reacts with any free amine in the protein, including the epsilon amino group of lysine. The amino group of the protein is linked to the aromatic ring of the DNB through an amine and to the dansyl group by a sulfonamide, and are hence stable to hydrolysis. The protein is hydrolyzed in 6 N HCl, and the amino acids separated by TLC or HPLC. Two spots should result if the protein was a single chain, with some Lys residues. The labeled amino acid other than Lys is the N-terminal amino acid. The C-terminal amino acid can be determined by addition of carboxypeptidases, enzymes which cleave amino acids from the C-terminal. A time course must be done to see which amino acid is released first. N-terminal analysis can also be done as part of sequencing the entire protein as discussed below (Edman degradation reaction).
Analysis for Specific Amino Acids
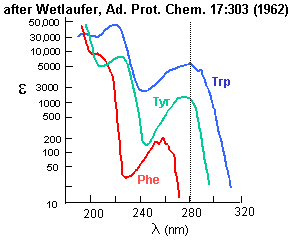
Aromatic amino acids can be detected by their characteristic absorbance profiles. Amino acids with specific functional groups can be determined by chemical reactions with specific modifying groups, as shown in section 2A.
Figure: amino acid absorbance profiles
Amino Acid Sequence - Edman Degradation
Two methods exist to determine the entire sequence of a protein. In one, the protein is sequenced; in the other, the DNA encoding the protein is sequenced, from which the amino acid sequence can be derived. The actually protein can be sequenced by automated, sequential Edman Degradation.
Figure: Edman Degradation
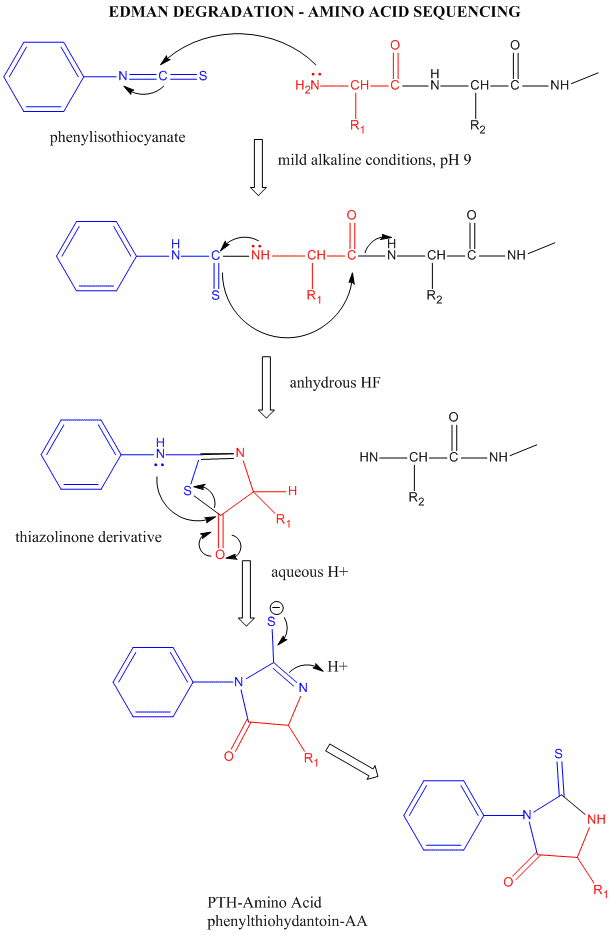
In this technique, a protein adsorbed to a solid phase reacts with phenylisothiocyanate. An intramolecular cyclization and cleavage of the N-terminal amino acid results, which can be washed from the adsorbed protein and detected by HPLC analysis. The yields in this technique are close to 100%. However, with time, more chains accumulate in which an N-terminal amino acid has not been removed. If it is removed on the next step, two amino acids will elute, creating increasing "noise" in the elution step - i.e. more than 1 amino acid derivative will be detected. Hence the maximal length of the peptide which can be sequenced is about 50 amino acids. Most proteins are larger than that. Hence, before the protein can be sequenced, it must be cleaved with specific enzymes called endoproteases which cleave proteins after specific side chains. For example, trypsin cleaves proteins within a chain after Lys and Arg, while chymotrypsin cleaves after aromatic amino acids, like Trp, Tyr, and Phe. Chemical cleavage by small molecules can be used as well. Cyanogen bromide, CNBr, cleaves proteins after methionine side chains. The individual proteins must be cleaved using two different methods, and each peptide fragment isolated and sequenced. Then the order of the cleaved peptides with known sequence can be pieced together by comparing the peptide sequences obtained using different cleavage methods. Many proteins also have disulfide bonds connecting Cys side chains distal to each other in the polypeptide chain. Proteolytic or chemical cleavage of the protein would lead to the formation of a fragment containing two peptides linked by disulfides. Edman degration would release two amino acids from such fragments. To avoid this problem, the protein is oxidized with performic acid, which irreversibly oxidizes free Cys, or Cys-Cys disulfides to cysteic acid residues. A summary of the steps involved in protein sequencing are shown below:
PROTEIN SEQUENCING STRATEGY - 8 STEPS
- If the protein contains more than one polypeptide chain, the chains are separated and purified. If disulfide bonds connect two different chains, the S-S bond must be cleaved (as described in step 2) and each peptide independently purified.
- Intrachain S-S bonds between Cys side chains are cleaved with performic acid. (See above for interchain S-S bonds).
- The amino acid composition of each chain is determined
- The N-terminal and C-terminal residues are identified.
- Each polypeptide chain is cleaved into smaller fragments, and the amino acid composition and sequence of each fragment is determined.
- Step 5 is repeated, using a different cleavage procedure to generate a different and overlapping set of peptide fragments.
- The overall amino acid sequence of the protein is reconstructed from the sequences in overlapping fragments.
- The position of the S-S is located. (See online problem set - Proteins)