
C11. Conformational and Binding - Examples
- Page ID
- 4973

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Conformational changes in macromolecule structure upon ligand binding are common features of such interactions. Interesting examples of conformational effects on binding are described below. Protein activities can often be modeled using the assumption of two major, different conformational forms. The first example, however, describes time-resolved structural changes in a protein on ligand release.
Unbinding of a ligand from a protein
As a complete understanding of an organic reaction requires a knowledge of the structures of reactants, transition states, intermediates and products along the reaction coordinates, so is the knowledge of different structural states along the dissociative pathway necessary for a complete understanding of the dissociative (and by inference the associative) mechanisms of ligand binding. Such a time-resolved pathway can be studied through molecular dynamics simulations. Schotte et al. have recently studied the dissociation of CO from carboxy-L29F-myoglobin to deoxy-L29F-Mb using time-resolved x-ray crystallography. (L29F is a mutant form of the wild type in which Leu 29 has been changed to Phe.) They chose this mutant since evidence of a short-lived (140 ps lifetime) intermediate in the flash-photolyzed dissociation of CO from the mutant was found using time-resolved mid-infrared spectroscopy of CO-L29F-myoglobin. In this technique, the CO-L29F-Mb is pulsed with an orange laser to photolyse the CO-Mb complex. After a brief delay, an IR pulse tuned to the CO stretch was added. The spectra of the mutant Mb showed two bands which converted to one broad band with time. The two bands represent two different orientations of the CO at the heme Fe, while the broad band arises when the CO dissociates from the primary site to other sites. The intermediate represented a structure in which CO was trapped in a binding site adjacent to the heme Fe which presumably is also a binding site for the initial association of the ligand. This site found in molecular dynamics simulations consisted of the heme and Val68, Ile 107, and Leu29 side chains, all of which are conserved in mammalian myoglobins. Time-resolved x-ray structures were obtained after orange laser-induced photolysis, followed by time-delayed x-ray pulses. These structures showed much larger changes in structure than the differences in structure between the deoxy-and oxy-Mb x-ray structures would suggest. The side changes exhibits coordinated motions that "sweeps" the CO away, accounting for the fast dissociation of the bound CO.
An Engineered Allosteric Enzyme
Do you ever wonder if your respiratory infection is viral, bacterial, or fungal in origin? Most patients would probably like an antibiotic but with the rise of resistant bacteria, unwarranted use of antibiotics is not wise. Wouldn't it be great if a quick test could be done to distinguish among these possibilities? A new sensitive and rapid method to analyze for specific DNA sequences (which could provide the needed distinction) has been developed by Saghatelian et. al. They have made an enzyme with a covalently attached single-stranded DNA sequence - an inhibitor-DNA-enzyme (IDE) complex.
Figure: inhibitor-DNA-enzyme (IDE) complex
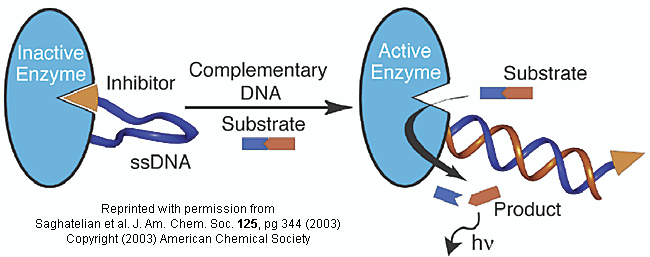
To the other end of the DNA is covalently attached an inhibitor of the enzyme. The inhibitor, tethered to the ssDNA, can bind to and inhibit the enzyme. Now if a complementary strand of DNA (derived from the bacterial, virus, etc) is added, it can bind to the ssDNA tether through complementary H-bond interactions to form a dsDNA in which the inhibitor is removed from the active site of the enzyme. The complementary DNA is, in effect, an allosteric activator, or more accurately a deinhibitor, of the enzyme. If a substrate is now added which can bind to the enzyme active site and form a fluorescent product, a very sensitive and quick assay is available. Saghatelian used a protease from B. cereus (CNP) and the substrate DABCYL-peptide-EDANS. The DABCYL group on this substrate is a quencher or the EDANS fluorophore. When cleaved by the enzyme, EDANS fluoresces intensely. The assay detected 10 fmol of DNA in less than three minutes.
Controlling the activity of a protein may be achieved through the binding of an allosteric effector at specific binding sites on a given protein. Another technique for allosteric control is to bind two monomeric proteins to form a two-domain complex in which one monomer allosterically activate the other. Lee et al recently engineering such a binding interaction using a light-sensing protein, phototropin LOV2 (LOV2), as the allosteric effector domain and E. coli dihydrofolate reductase (EcDHFR) as the catalytical domain. The reaction is monitored by the reduction of dihydofolate to tetrahydrafolate. Investigators made chimera in with the LOV2 domain was connected through its N and C terminal helices to bound to EcDHFR at two sites on its surface: Site A, which noticeably affected the activity of the EcDHFR active site, and Site B, which had little influence on the rate of hydride transfer, khyd (EcDHFR activity). The khyd of the chimeric LOV2-EcDHFR protein bound at Site B was not affected by the conformational shifts of the allosteric effector, which were induced by light. On the other hand, the khyd of the Site A protein was significantly enhanced when light modified the LOV2 structure. This successfully demonstrated substrate activity modification by adjusting the effector.
Circadian Rhythms and the Biological Clock
Many human behaviors are cyclic with a repeat period of 24 hours (such as sleep/wake cycles). These circadian rhythms, which can be phase-shifted by alternations in environmental cues (such as light intensity, sleep deprivation) must have some biochemical basis. Understanding the mechanisms underlying circadian rhythms and its regulations would prove helpful in developing new ways to help people minimize the influence of jet lag or shift work. In humans, light intensity information is transmitted from the retina to the suprachiasmatic nucleus in the hypothalmus (in the brain), which then signals the pineal gland (behind the hypothalmus) to secrete the hormone melatonin. Its levels rise at night and fall in the day, but its biochemical mechanism of action is still being determined. Other hormones are also involved. What are the biochemical targets of these hormones? What is the basis of circadian changes in individual cells?
One such protein is the membrane enzyme (Hydroquinone) NADH Oxidase (NOX). NADH is a small cellular reducing agent that we will discuss in the future. This enzyme is found in the external plasma membrane of all human cells, including tumor cells. NOX proteins are very usual in that they possess two activities: a NADH oxidase activity (measured by the disappearance of of NADH) and a protein disulfide isomerase (thiol interchange) activity (measured by the renaturation of RNase A using cCMP as a substrate or the cleavage of dithiodipyridine). These activities alternate in a temporal sense with a 24 minute period! The normal cellular form of the protein is constitutively expressed and responsive to hormones. A form found specifically on tumor cells, tNOX, is inhibited by certain chemotherapeutic drugs and by capsaicin (the active ingredient in hot peppers), and is not responsive to hormones (suggesting unregulated activity). tNOX has a 22 minute period. In cancer patients, a truncated form of tNOX (ttNOX), formed by limited proteolysis from tNOX, is found in the serum. NOX proteins are also resistant to proteases and can self aggregate to form "amyloid" type fibrils similar to those found in prion diseases. They can also interact with other proteins and render them protease resistant. The temporal periods of both proteins are independent of temperature and can be "entrained" through the appropriate stimuli. What structural features of the protein can account for unique properties. Early evidence (using FTIR and CD) suggests that changes in secondary structure (similar to prion proteins) occurs involving changes from alpha helices to beta sheets. Perhaps the protein can exist in two distinct, yet similar conformations, each with a different activity.
The tNOX gene has been cloned and expressed. It encodes a protein of 610 amino acids, and its activities, when expressed in bacteria, cycle in 22 minutes. Site-specific mutations produce periods of 36 minutes (for Cys575Ala) and 42 minutes (for Cys558Ala) mutants. These proteins show the same activities and periods when produced in transformed eukaryotic COS-1 cells. This cell surface proteins affects the circadian rhythms of the whole cell. A normal protein in the cell, glyceraldehyde -3-phosphate dehydrogenase, (GAPDH) exhibits a 24 hour activity circadian rhythm in normal COS cells (which have a constitutively expressed NOX gene product). When transformed with the mutants, the demonstrate not only a 24 hour GAPDH activity, but in addition, a 22, 36, or 42 hour activity when transformed with the gene for tNOX, the tNOX Cys575Ala mutant, and the tNOX Cys558Ala mutant respectively.
HIV Binding to T helper immune cells
It has been notoriously difficult to develop a vaccine against the HIV virus. One type of vaccine results in the formation of protective proteins called antibodies, which bind to an immunogen in the vaccine and ultimately to a "foreign" molecule such as a protein on the surface of an actual virus or bacterial cell. The HIV has a surface protein, gp120, which binds to a receptor protein, CD4, on the surface of certain immune cells like the T helper cell. If this interaction could be prevented, then HIV could not enter cells. Many attempts have been made to develop neutralizing antibodies to gp120 by using gp120 and variants as an immunogen. These have all failed. Further work has shown that when gp120 binds to the receptor protein CD4, the gp120 undergoes a conformational change which allows a newly exposed loop on gp120 to bind to yet another coreceptor, CCR5, on the immune cell, facilitating virus uptake into the cell. The exposed loop conformation of the gp120 seems to resemble the structure of a normal protein ligand (the chemokine RANTES) for the CCR5 receptor. Such examples of molecular mimicry are becoming more common. Recently, the crystal structure of an unliganded simian virus gp120 protein has been determined (Chen et al.). Comparison of this to the bound form (to CD4) shows clear conformational changes. Small inhibitors could be designed to the unbound form, locking it in that conformation which would prevent viral entry into cells.
First consider the cell bound state. There appears to be a very large structural change in gp120 on binding CD4. When this happens, another part of the gp120 protein is exposed, which then binds to another protein "coreceptor" on the cell. This dual binding to the CD4 and the coreceptor "hides" the gp120 from potential neutralizing antibodies, perhaps by steric exclusion.
What about the fee state? Why can't it interact with neutralizing antibodies? Chen et al have recently determined the structure of the free gp120 protein. On binding to CD4, half of the gp120 refolds, bringing distal residues together to allow binding to co-receptor. Before binding, three copies of gp120 are found on surface viral spikes. Critical residues on gp120 for possible immune recognition appear to be inhibited from antibody binding since they point towards each other which sequesters them. In addition, carbohydrates covalently attached to the gp120 also shield amino acid side chains from immune recognition. Such large changes in protein structure are also seen with part of the influenza virus surface proteins that allow fusion of viral and target cell membranes, and subsequent viral uptake into the cell. They may be a hallmark of viral-target cell interactions.
To a first approximation, the protein may be considered to have two formations, the free and bound forms. Any small molecule which would preferentially bind to the free form could shift the equibrium to that from, and hence prevent viral infectivity. Antibodies that have those properties (i.e. neutralizing) appear to be difficult for the immune system to make. However, small ligand which bind in cavities present in the free form would have such properties. New viral-entry inhibitors have been found that appear to bind in such cavities. Lin et al. PNAS 100, 11013 (2003)
 Jmol: Updated Unliganded gp120 core (HIV viral protein) ) Jmol14 (Java) | JSMol (HTML5) NOTUPDATED
Jmol: Updated Unliganded gp120 core (HIV viral protein) ) Jmol14 (Java) | JSMol (HTML5) NOTUPDATED
Parainfluenza Virus Protein
The parainfluenza 5F protein catalyzes the viral and cellular membranes allowing for the entry of the viral genome into the cell. The F protein undergoes refolding during this process, which leads to differing conformations when it is in the pre- and post- transitional states. The parainfluenza virus is similar to other paramyxovirides, (enveloped viruses) which include: mumps, measles, sendai, Newcastle, and the human respiratory sincytial virus. The parainfluenza 5F protein consists of a globular head attached to a trimeric coiled-coil stalk formed by the C-terminal of the HRB region. This exact form of the virus shown in this tutorial is that of the pre-conformational state in which the stalk sits above the viral membrane.
Jmol: Updated Parainfluenza Virus 5F Protein Jmol14 (Java) | JSMol (HTML5)
Additional Links
-
 PDB - Binding Database: A comprehensive collection of the experimentally measured binding affinity data for all types of biomolecular complexes deposited in the Protein Data Bank (PDB).
PDB - Binding Database: A comprehensive collection of the experimentally measured binding affinity data for all types of biomolecular complexes deposited in the Protein Data Bank (PDB). - Protein Interaction Domains
- Jmol:Examples of Jmol Ligand/Protein Complexes