
W2018_Bis2A_Lecture20_reading
- Page ID
- 10818
Replication: proofreading
When the cell begins the task of replicating the DNA, it does so in response to environmental signals that tell the cell it is time to divide. The ideal goal of DNA replication is to produce two identical copies of the double-stranded DNA template and to do it in an amount of time that does not pose an unduly high evolutionarily selective cost. This is a daunting task when you consider that there are ~6,500,000,000 base pairs in the human genome and ~4,500,000 base pairs in the genome of a typical E. coli strain and that Nature has determined that the cells must replicate within 24 hours and 20 minutes, respectively. In either case, many individual biochemical reactions need to take place.
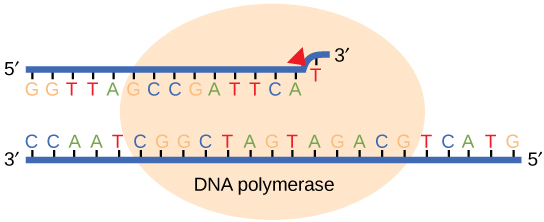
While ideally replication would happen with perfect fidelity, DNA replication, like all other biochemical processes, is imperfect—bases may be left out, extra bases may be added, or bases may be added that do not properly base-pair. In many organisms, many of the mistakes that occur during DNA replication are promptly corrected by DNA polymerase itself via a mechanism known as proofreading. In proofreading, the DNA polymerase "reads" each newly added base via sensing the presence or absence of small structural anomalies before adding the next base to the growing strand. In doing so, a correction can be made.
If the polymerase detects that a newly added base has paired correctly with the base in the template strand, the next nucleotide is added. If, however, a wrong nucleotide is added to the growing polymer, the misshaped double helix will cause the DNA polymerase to stall, and the newly made strand will be ejected from the polymerizing site on the polymerase and will enter into an exonuclease site. In this site, DNA polymerase is able to cleave off the last several nucleotides that were added to the polymer. Once the incorrect nucleotides have been removed, new ones will be added again. This proofreading capability comes with some trade-offs: using an error-correcting/more accurate polymerase requires time (the trade-off is speed of replication) and energy (always an important cost to consider). The slower you go, the more accurate you can be. Going too slow, however, may keep you from replicating as fast as your competition, so figuring out the balance is key.
Errors that are not corrected by proofreading become what are known as mutations.
Suggested discussion
Why would DNA replication need to be fast? Consider the environment the DNA is in, and compare that to the structure of DNA while being replicated.
Suggested discussion
What are the pros and cons of DNA polymerase's proofreading capabilities?
Replication mistakes and DNA repair
Although DNA replication is typically a highly accurate process, and proofreading DNA polymerases helps to keep the error rate low, mistakes still occur. In addition to errors of replication, environmental damage may also occur to the DNA. Such uncorrected errors of replication or environmental DNA damage may lead to serious consequences. Therefore, Nature has evolved several mechanisms for repairing damaged or incorrectly synthesized DNA.
Mismatch repair
Some errors are not corrected during replication but are instead corrected after replication is completed; this type of repair is known as a mismatch repair. Specific enzymes recognize the incorrectly added nucleotide and excise it, replacing it with the correct base. But, how do mismatch repair enzymes recognize which of the two bases is the incorrect one?
In E. coli, after replication, the nitrogenous base adenine acquires a methyl group; this means that directly after replication the parental DNA strand will have methyl groups, whereas the newly synthesized strand lacks them. Thus, mismatch repair enzymes are able to scan the DNA and remove the wrongly incorporated bases from the newly synthesized, non-methylated strand by using the methylated strand as the "correct" template from which to incorporate a new nucleotide. In eukaryotes, the mechanism is not as well understood, but it is believed to involve recognition of unsealed nicks in the new strand, as well as a short-term, continuing association of some of the replication proteins with the new daughter strand after replication has completed.
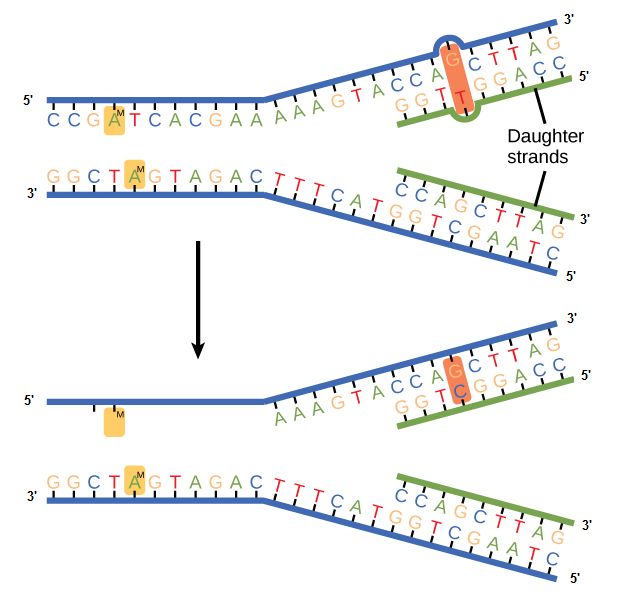
Figure 2. In mismatch repair, the incorrectly added base is detected after replication. The mismatch repair proteins detect this base and remove it from the newly synthesized strand by nuclease action. The gap is now filled with the correctly paired base.
Nucleotide excision repair
Nucleotide excision repair enzymes replace incorrect bases by making a cut on both the 3' and 5' ends of the incorrect base. The entire segment of DNA is removed and replaced with correctly paired nucleotides by the action of a DNA polymerase. Once the bases are filled in, the remaining gap is sealed with a phosphodiester linkage catalyzed by the enzyme DNA ligase. This repair mechanism is often employed when UV exposure causes the formation of pyrimidine dimers.
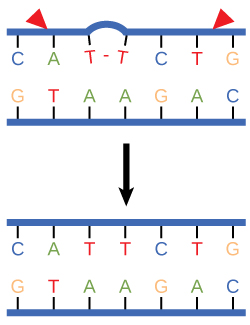
Figure 3. Nucleotide excision repairs thymine dimers. When exposed to UV, thymines lying adjacent to each other can form thymine dimers. In normal cells, they are excised and replaced.
Consequences of errors in replication, transcription, and translation
Something key to think about:
Cells have evolved a variety of ways to make sure DNA errors are both detected and corrected. We have already discussed several of them. But why did so many different mechanisms evolve? From proofreading by the various DNA-dependent DNA polymerases, to the complex repair systems. Such mechanisms did not evolve for errors in transcription or translation. If you are familiar with the processes of transcription and/or translation, think about what the consequences would be of an error in transcription. Would such an error affect the offspring? Would it be lethal to the cell? What about errors in translation? Ask the same questions about the process of translation. What would happen if the wrong amino acid is accidentally put into the growing polypeptide during translation? How do these contrast with DNA replication? If you are not familiar with transcription or translation, don't fret. We'll learn those soon and return to this question again.
Genomes as organismal blueprints
A genome, not to be confused with a gnome, is an organism's complete collection of heritable information stored in DNA. Differences in information content help to explain the diversity of life we see all around us. Changes to the information encoded in the genome are the primary drivers of the phenotypic diversity we see (and some we can't) around us that are filtered by natural selection, and they are thus the drivers of evolution. This leads to questions. If every cell in a multicellular organism contains the same sequence of DNA, how can there be different cell types (e.g., how can a cell in a liver be so different from a cell in the brain if they both carry the same DNA)? How do we read the information? How do we interpret what we read? How do we understand how all of the "parts" we identify in the genome functionally interrelate? How is all of this related to the expression of traits? How do changes in the genome lead to changes in traits?
Determining a genome sequence
The information encoded in genomes provides important data for understanding life, its functions, its diversity, and its evolution. Therefore, it stands to reason that a reasonable place to begin studies in biology would be to read the information content encoded in the genome(s) in question. A good starting point is to determine the sequence of nucleotides (A, G, C, T) and their organization into one or more independently replicating units of DNA (e.g., think chromosomes and/or plasmids ). For 30+ years after the discovery that DNA is the hereditary material, this was a daunting proposition. In the late 1980s, however, the advent of semi-automated tools for DNA sequencing were pioneered, and this began a revolution that has dramatically changed how we approach the study of life. Twenty years later, in the mid-2000s, we entered a period of accelerated technological progress in which advances in materials sciences (particularly, advances in our ability to make things on a very small scale), optics, electrical and computer engineering, bioengineering, and computer sciences have all converged to bring us dramatic increases in our capacity to sequence DNA and correspondingly dramatic decreases in the cost of numerous advances in our ability to sequence DNA. A famous example to illustrate this point is to compare the changes in cost to sequence the human genome. The first draft of the human genome took nearly 15 years and $3 billion dollars to complete. Today, 10's of human genomes can be sequenced in a single day on a single instrument at a cost of less than $1000 each (the cost and time continue to decrease). Today, companies like Illumina, Pacific Biosciences, Oxford Nanopore, and others offer competing technologies that are driving down the cost and increasing the volume, quality, speed, and portability of DNA sequencing.
One of the very exciting elements of the DNA sequencing revolution is that it has required and continues to require contributions from biologists, chemists, materials scientists, electrical engineers, mechanical engineers, computer scientists and programmers, mathematicians and statisticians, product developers, and many other technical experts. The potential applications and implications of unlocking barriers to DNA sequencing have also engaged investors, business people, product developers, entrepreneurs, ethicists, policy makers, and many others to pursue new opportunities and to think about how to best and most responsibly use this growing technology.
The technological advances in genome sequencing have resulted in a virtual flood of complete genome sequences being determined and deposited into publicly available databases. You can find many of them at the National Center for Biotechnology Information. The number of available , completely sequenced genomes numbers in the tens of thousands—over 2,000 eukaryotic genomes, over 600 archaeal genomes, and nearly 12,000 bacterial genomes at the time of this writing. Tens of thousands of more genome sequencing projects are in progress. With this many genome sequences available—or soon to be available—we can start asking many questions about what we see in these genomes. What patterns are common to all genomes? How many genes are encoded in genomes? How are these organized? How many different types of features can we find? What do the features that we find do? How different are the genomes from one another? Is there evidence that can tell us how genomes evolve? Let's briefly examine a few of these questions.
Diversity of genomes
Diversity of sizes, number of genes, and chromosomes
Let's start by examining the range of genome sizes. In the table below, we see a sampling of genomes from the database. We can see that the genomes of free living organisms range tremendously in size. The smallest known genome is encoded in 580,000 base pairs while the largest is 150 billion base pairs—for reference, recall that the human genome is 3.2 billion base pairs. That's a huge range of sizes. Similar disparities in the number of genes also exist.
Table 1. This table shows some genome data for various organisms. 2n = diploid number. Attribution: Marc T. Facciotti (own work—reproduced from http://book.bionumbers.org/how-big-are-genomes/)

Examining Table 1 also reveals that some organisms carry with them more than one chromosome. Some genomes are also polyploid, meaning that they maintain multiple copies of similar but not identical (homologous) copies of each chromosome. A diploid organism carries in its genome two homologous copies (usually one from Mom and one from Dad) of each chromosome. Humans are diploid. Our somatic cells carry 2 homologous copies of 23 chromosomes. We received 23 copies of individual chromosomes from our mother and 23 copies from our father, for a total of 46. Some plants have higher ploidy. For example, a plant with four homologous copies of each chromosome is termed tetraploid. An organism with a single copy of each chromosome is termed haploid.
Structure of genomes
Table 1 also provides clues to other points of interest. For instance, if we compare the pufferfish genome to the chimpanzee genome, we note that they encode roughly the same number of genes (19,000), but they do so on dramatically differently sized genomes—400 million base pairs versus 3.3 billion base pairs, respectively. That implies that the pufferfish genome must have much less space between its genes than what might be expected to be found in the chimpanzee genome. Indeed, this is the case, and the difference in gene density is not unique to these two genomes. If we look at Figure 1, which attempts to represent a 50-kb part of the human genome, we notice that in addition to the protein-coding regions (indicated in red and pink) that many other so-called "features" can be read from the genome. Many of these elements contain highly repetitive sequences.

Figure 1. This figure shows a 50-kb segment of the human β T-cell receptor locus on chromosome 7. This figure depicts a small region of the human genome and the types of "features" that can be read and decoded in the genome, including, but also in addition to, protein-coding sequences. Red and pink correspond to regions that encode proteins. Other colors represent different types of genomic elements. Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
If we now look at what fraction of the whole human genome each of these types of elements makes up (see Figure 2), we see that protein-coding genes only make up 48 million of the 3.2 billion bases of the haploid genome.

Figure 2. This graph depicts how the many base pairs of DNA in the human haploid genome are distributed between various identifiable features. Note that only a small fraction of the genome is associated directly with protein-coding regions. Attribution: Marc T. Facciotti (own work—reproduced from sources noted in figure)
When we examine the frequency of repeat regions versus protein-coding regions in different species, we note large differences in protein-coding versus non-coding regions.

Figure 3. This figure shows 50-kb segments of different genomes, illustrating the highly variable frequency of repeat versus protein-coding elements in different species.
Attribution: Marc T. Facciotti (own work—reproduced from www.ncbi.nlm.nih.gov/books/NBK21134/)
Suggested discussion
Propose a hypothesis for why you think some genomes might have more or fewer noncoding sequences.
Dynamics of genome structure
Genomes change over time, and numerous different types of events can change their sequence.
1. Mutations are either accumulated during DNA replication or through environmental exposure to chemical mutagens or radiation. These changes typically occur at the level of single nucleotides.
2. Genome rearrangements describe a class of large-scale changes that can occur, and they include the following: (a) deletions—where segments of the chromosome are lost; (b) duplication—where regions of the chromosome are inadvertently duplicated; (c) insertions—the insertion of genetic material (note that sometimes this is acquired from viruses or the environment, and deletion/insertion pairs may happen across chromosomes); (d) inversions—where regions of the genome are flipped within the same chromosome; and (e) translocations—where segments of the chromosome are translocated (moved elsewhere in the chromosome).
These changes happen at different rates, and some are facilitated by the activity of enzyme catalysts (e.g., transposases).
The study of genomes
Comparative genomics
One of the most common things to do with a collection of genome sequences is to compare the sequences of multiple genomes to one another. In general terms, these types of activities fall under the umbrella of a field called comparative genomics.
Comparing the genomes of people who suffer from an inheritable disease to the genomes of people who are not afflicted can help us to uncover the genetic basis for the malady. Comparing the gene content, order, and sequence of related microbes can help us find the genetic basis of why some microbes cause disease while their close cousins are virtually harmless. We can compare genomes to understand how a new species may have evolved. There are many possible analyses! The basis of these analyses is similar: look for differences across multiple genomes and try to associate those differences with different traits or behaviors in those organisms.
Lastly, some people are comparing genome sequences to try to understand the evolutionary history of the organisms. Typically, these types of comparisons result in a graph known as a phylogenetic tree, which is a graphical model of the evolutionary relationship between the various species being compared. This field, not surprisingly, is called phylogenomics.
Metagenomics: who is living somewhere and what are they doing?
In addition to studying the genomes of individual species, the increasingly powerful DNA-sequencing technologies are making it possible to simultaneously sequence the genomes of environmental samples that are inhabited by many different species. This field is called metagenomics. These studies are typically focused on trying to understand what microbial species inhabit different environments. There is great interest in using DNA sequencing to study the populations of microbes in the gut and to watch how the population changes in response to different diets, to see if there is any association between the abundance of different microbes and various diseases, or to look for the presence of pathogens. People are using DNA sequencing of environmental metagenomic samples to explore which microbes inhabit different environments on Earth (from the deep sea, to soil, to air, to hypersaline ponds, to cat feces, to some of the common surfaces we touch every day).
In addition to discovering "who lives where," the sequencing of microbial populations in different environments can also reveal what protein-coding genes are present in an environment. This can give investigators clues into what metabolic activities might be occurring in that environment. In addition to providing important information about what kind of chemistry might be happening in a specific environment, the catalog of genes that is accumulated can also serve as an important resource for the discovery of novel enzymes for applications in biotechnology.
The flow of genetic information
In bacteria, archaea, and eukaryotes, the primary role of DNA is to store heritable information that encodes the instruction set required for creating the organism in question. While we have gotten much better at quickly reading the chemical composition (the sequence of nucleotides in a genome and some of the chemical modifications that are made to it), we still don't know how to reliably decode all of the information within and all of the mechanisms by which it is read and ultimately expressed.
There are, however, some core principles and mechanisms associated with the reading and expression of the genetic code whose basic steps are understood and that need to be part of the conceptual toolkit for all biologists. Two of these processes are transcription and translation, which are the coping of parts of the genetic code written in DNA into molecules of the related polymer RNA and the reading and encoding of the RNA code into proteins, respectively.
In BIS2A, we focus largely on developing an understanding of the process of transcription (recall that an Energy Story is simply a rubric for describing a process) and its role in the expression of genetic information. We motivate our discussion of transcription by focusing on functional problems (bringing in parts of our problem solving/design challenge rubric) that must be solved the the process to take place. We then go on to describe how the process is used by Nature to create a variety of functional RNA molecules (that may have various structural, catalytic or regulatory roles) including so called messenger RNA (mRNA) molecules that carry the information required to synthesize proteins. Likewise, we focus on challenges and questions associated with the process of translation, the process by which the ribosomes synthesize proteins.
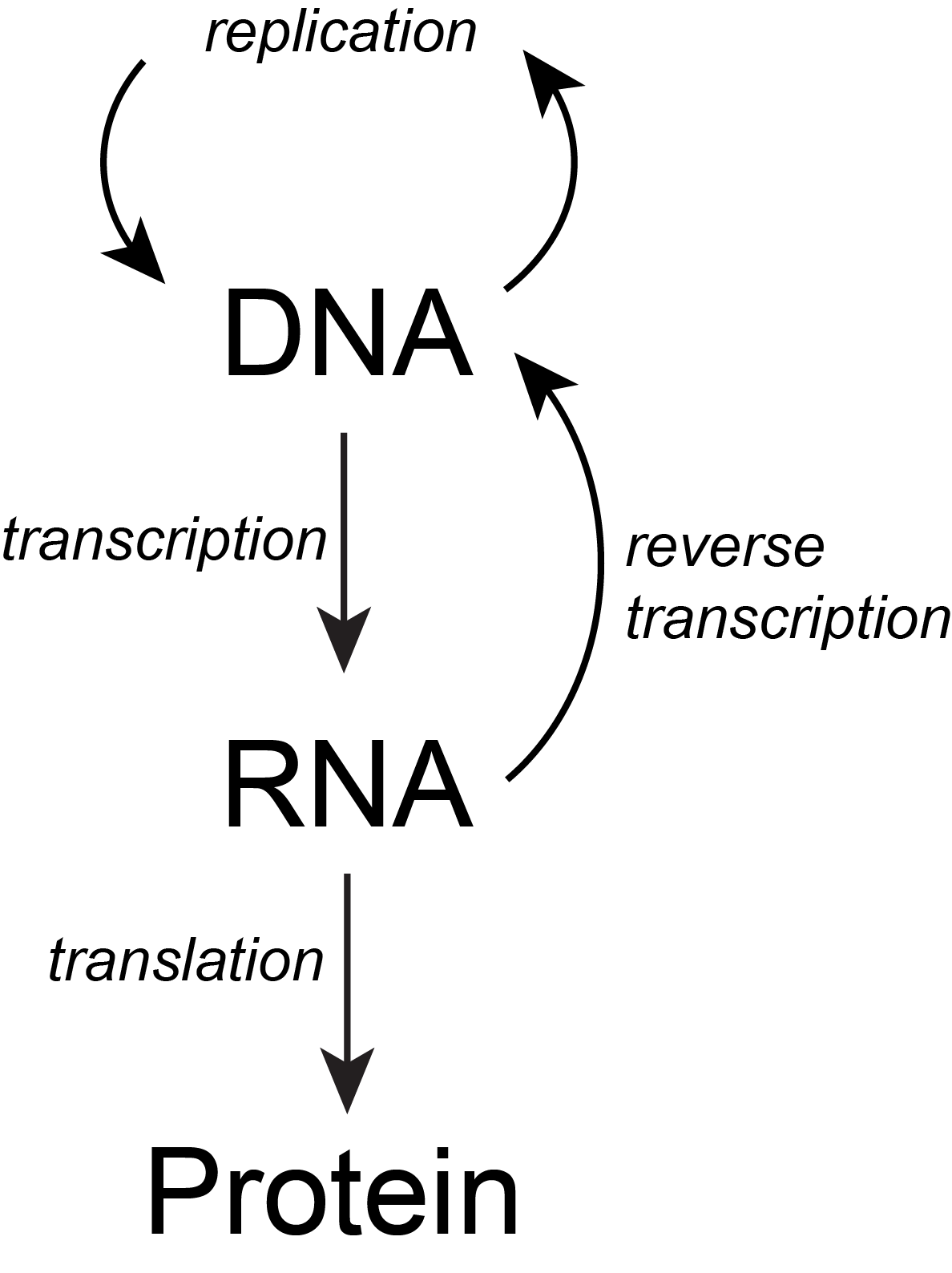
The basic flow of genetic information in biological systems is often depicted in a scheme known as "the central dogma" (see figure below). This scheme states that information encoded in DNA flows into RNA via transcription and ultimately to proteins via translation. Processes like reverse transcription (the creation of DNA from and RNA template) and replication also represent mechanisms for propagating information in different forms. This scheme, however, doesn't say anything per se about how information is encoded or about the mechanisms by which regulatory signals move between the various layers of molecule types depicted in the model. Therefore, while the scheme below is a nearly required part of the lexicon of any biologist, perhaps left over from old tradition, students should also be aware that mechanisms of information flow are more complex (we'll learn about some as we go, and that "the central dogma" only represents some core pathways).
Genotype to phenotype
An important concept in the following sections is the relationship between genetic information, the genotype, and the result of expressing it, the phenotype. These two terms and the mechanisms that link the two will be discussed repeatedly over the next few weeks—start becoming proficient with using this vocabulary.

Figure 2. The information stored in DNA is in the sequence of the individual nucleotides when read from 5' to 3' direction. Conversion of the information from DNA into RNA (a process called transcription) produces the second form that information takes in the cell. The mRNA is used as the template for the creation of the amino acid sequence of proteins (in translation). Here, two different sets of information are shown. The DNA sequence is slightly different, resulting in two different mRNAs produced, followed by two different proteins, and ultimately, two different coat colors for the mice.
Genotype refers to the information stored in the DNA of the organism, the sequence of the nucleotides, and the compilation of its genes. Phenotype refers to any physical characteristic that you can measure, such as height, weight, amount of ATP produced, ability to metabolize lactose, response to environmental stimuli, etc. Differences in genotype, even slight, can lead to different phenotypes that are subject to natural selection. The figure above depicts this idea. Also note that, while classic discussions of the genotype and phenotype relationships are talked about in the context of multicellular organisms, this nomenclature and the underlying concepts apply to all organisms, even single-celled organisms like bacteria and archaea.
Note: possible discussion
Can something you can not see "by eye" be considered a phenotype?
Note: possible discussion
Can single-celled organisms have multiple simultaneous phenotypes? If so, can you propose an example? If not, why?
Genes
What is a gene? A gene is a segment of DNA in an organism's genome that encodes a functional RNA (such as rRNA, tRNA, etc.) or protein product (enzymes, tubulin, etc.). A generic gene contains elements encoding regulatory regions and a region encoding a transcribed unit.
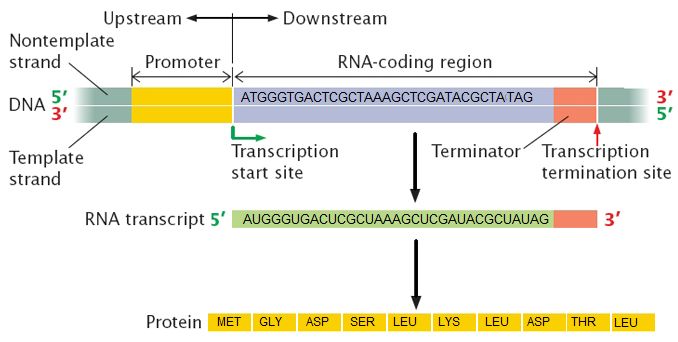
Genes can acquire mutations—defined as changes in the in the composition and or sequence of the nucleotides—either in the coding or regulatory regions. These mutations can lead to several possible outcomes: (1) nothing measurable happens as a result; (2) the gene is no longer expressed; or (3) the expression or behavior of the gene product(s) are different. In a population of organisms sharing the same gene different variants of the gene are known as alleles. Different alleles can lead to differences in phenotypes of individuals and contribute to the diversity in biology that is under selective pressure.
Start learning these vocabulary terms and associated concepts. You will then be somewhat familiar with them when we start diving into them in more detail over the next lectures.