
1.52: Genomics*
- Page ID
- 21299

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Genomics
The study of nucleic acids began with the discovery of DNA, progressed to the study of genes and small fragments, and has now exploded to the field of genomics. Genomics is the study of entire genomes, including the complete set of genes, their nucleotide sequence and organization, and their interactions both within a species and with other species. The advances in genomics have been made possible by DNA sequencing technology. Just as information technology has led to Google Maps, enabling us to get detailed information about locations around the globe, genomic information is used to create similar maps of the DNA of different organisms.
Mapping genomes
Genome mapping is the process of finding the location of genes on each chromosome. The maps that are created are comparable to the maps that we use to navigate streets. A genetic map is an illustration that lists genes and their location on a chromosome. Genetic maps provide the big picture (similar to a map of interstate highways) and use genetic markers (similar to landmarks). A genetic marker is a gene or sequence on a chromosome that shows genetic linkage with a trait of interest. The genetic marker tends to be inherited with the gene of interest. One measure of distance between them is the recombination frequency during meiosis; early geneticists called this linkage analysis.
Physical maps get into the intimate details of smaller regions of the chromosomes (similar to a detailed road map). A physical map is a representation of the physical distance, in nucleotides, between genes or genetic markers. Both genetic linkage maps and physical maps are required to build a complete picture of the genome. Having a complete map of the genome makes it easier for researchers to study individual genes. Human genome maps help researchers in their efforts to identify human disease-causing genes related to illnesses such as cancer, heart disease, and cystic fibrosis, to name a few. In addition, genome mapping can be used to help identify organisms with beneficial traits, such as microbes with the ability to clean up pollutants or even prevent pollution. Research involving plant genome mapping may lead to agricultural methods that produce higher crop yields or to the development of plants that adapt better to climate change.
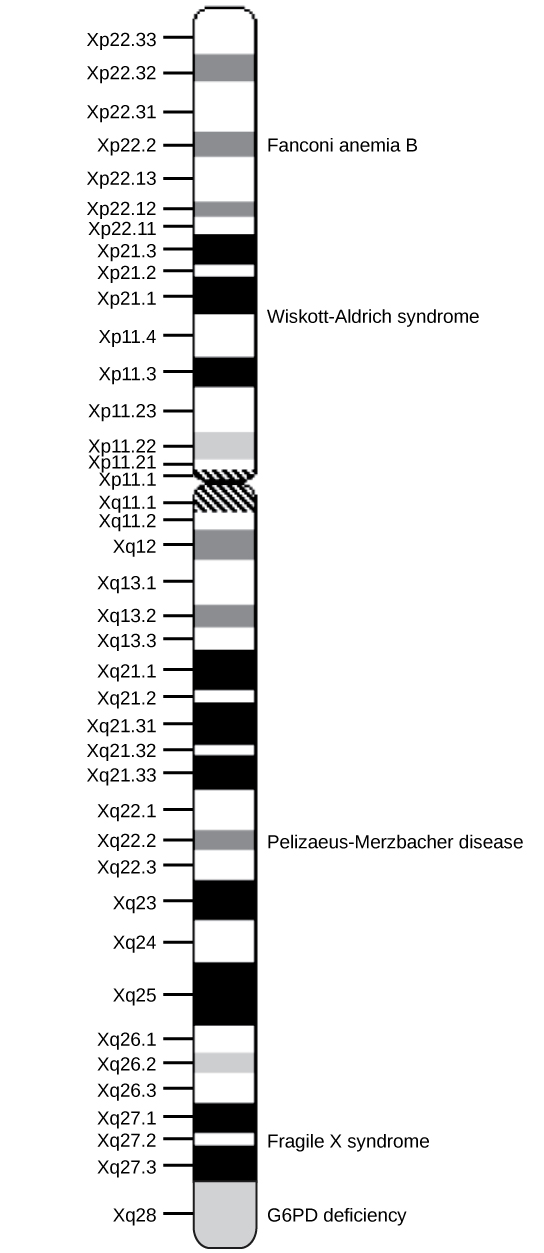
Figure 1. This is a physical map of the human X chromosome. Credit: modification of work by NCBI, NIH
Genetic maps provide the outline, and physical maps provide the details. It is easy to understand why both types of genome-mapping techniques are important to show the big picture. Information obtained from each technique is used in combination to study the genome. Genomic mapping is used with different model organisms that are used for research. Genome mapping is still an ongoing process, and as more advanced techniques are developed, more advances are expected. Genome mapping is similar to completing a complicated puzzle using every piece of available data. Mapping information generated in laboratories all over the world is entered into central databases, such as the National Center for Biotechnology Information (NCBI). Efforts are made to make the information more easily accessible to researchers and the general public. Just as we use global positioning systems instead of paper maps to navigate through roadways, NCBI allows us to use a genome viewer tool to simplify the data mining process.
Whole genome sequencing
Although there have been significant advances in the medical sciences in recent years, doctors are still confounded by many diseases, and researchers are using whole genome sequencing to get to the bottom of the problem. Whole genome sequencing is a process that determines the DNA sequence of an entire genome. Whole genome sequencing is a brute-force approach to problem solving when there is a genetic basis at the core of a disease. Several laboratories now provide services to sequence, analyze, and interpret entire genomes.
In 2010, whole genome sequencing was used to save a young boy whose intestines had multiple mysterious abscesses. The child had several colon operations with no relief. Finally, a whole genome sequence revealed a defect in a pathway that controls apoptosis (programmed cell death). A bone marrow transplant was used to overcome this genetic disorder, leading to a cure for the boy. He was the first person to be successfully diagnosed using whole genome sequencing.
The first genomes to be sequenced, such as those belonging to viruses, bacteria, and yeast, were smaller in terms of the number of nucleotides than the genomes of multicellular organisms. The genomes of other model organisms, such as the mouse (Mus musculus), the fruit fly (Drosophila melanogaster), and the nematode (Caenorhabditis elegans) are now known. A great deal of basic research is performed in model organisms because the information can be applied to other organisms. A model organism is a species that is studied as a model to understand the biological processes in other species that can be represented by the model organism. For example, fruit flies are able to metabolize alcohol like humans, so the genes affecting sensitivity to alcohol have been studied in fruit flies in an effort to understand the variation in sensitivity to alcohol in humans. Having entire genomes sequenced helps with the research efforts in these model organisms.

Figure 2. Much basic research is done with model organisms, such as the mouse, Mus musculus; the fruit fly, Drosophila melanogaster; the nematode, Caenorhabditis elegans; the yeast, Saccharomyces cerevisiae; and the common weed, Arabidopsis thaliana. Credit: "mouse": modification of work by Florean Fortescuecredit; "nematodes": modification of work by "snickclunk"/Flickr; "common weed": modification of work by Peggy Greb, USDA; scale-bar data from Matt Russell
The first human genome sequence was published in 2003. The number of whole genomes that have been sequenced steadily increases and now includes hundreds of species and thousands of individual human genomes.
Applying genomics
The introduction of DNA sequencing and whole genome sequencing projects, particularly the Human Genome Project, has expanded the applicability of DNA sequence information. Genomics is now being used in a wide variety of fields, such as metagenomics, pharmacogenomics, and mitochondrial genomics. The most commonly known application of genomics is to understand and find cures for diseases.
Predicting disease risk at the individual level
Predicting the risk of disease involves screening and identifying currently healthy individuals by genome analysis at the individual level. Intervention with lifestyle changes and drugs can be recommended before disease onset. However, this approach is most applicable when the problem arises from a single gene mutation. Such defects only account for about five percent of diseases found in developed countries. Most of the common diseases, such as heart disease, are multifactorial or polygenic, which refers to a phenotypic characteristic that is determined by two or more genes, and also environmental factors such as diet. In April 2010, scientists at Stanford University published the genome analysis of a healthy individual (Stephen Quake, a scientist at Stanford University, who had his genome sequenced); the analysis predicted his propensity to acquire various diseases. A risk assessment was done to analyze Quake’s percentage of risk for 55 different medical conditions. A rare genetic mutation was found that showed him to be at risk for sudden heart attack. He was also predicted to have a 23 percent risk of developing prostate cancer and a 1.4 percent risk of developing Alzheimer’s disease. The scientists used databases and several publications to analyze the genomic data. Even though genomic sequencing is becoming more affordable and analytical tools are becoming more reliable, ethical issues surrounding genomic analysis at a population level remain to be addressed. For example, could such data be legitimately used to charge more or less for insurance or to affect credit ratings?
Genome-wide association studies
Since 2005, it has been possible to conduct a type of study called a genome-wide association study, or GWAS. A GWAS is a method that identifies differences between individuals in single nucleotide polymorphisms (SNPs) that may be involved in causing diseases. The method is particularly suited to diseases that may be affected by one or many genetic changes throughout the genome. It is very difficult to identify the genes involved in such a disease using family history information. The GWAS method relies on a genetic database that has been in development since 2002 called the International HapMap Project. The HapMap Project sequenced the genomes of several hundred individuals from around the world and identified groups of SNPs. The groups include SNPs that are located near eachother on chromosomes so they tend to stay together through recombination. The fact that the group stays together means that identifying one marker SNP is all that is needed to identify all the SNPs in the group. There are several million SNPs identified, but identifying them in other individuals who have not had their complete genome sequenced is much easier because only the marker SNPs need to be identified.
In a common design for a GWAS, two groups of individuals are chosen; one group has the disease, and the other group does not. The individuals in each group are matched in other characteristics to reduce the effect of confounding variables causing differences between the two groups. For example, the genotypes may differ because the two groups are mostly taken from different parts of the world. Once the individuals are chosen, and typically their numbers are a thousand or more for the study to work, samples of their DNA are obtained. The DNA is analyzed using automated systems to identify large differences in the percentage of particular SNPs between the two groups. Often the study examines a million or more SNPs in the DNA. The results of GWAS can be used in two ways: the genetic differences may be used as markers for susceptibility to the disease in undiagnosed individuals, and the particular genes identified can be targets for research into the molecular pathway of the disease and potential therapies. An offshoot of the discovery of gene associations with disease has been the formation of companies that provide so-called “personal genomics”, which will identify risk levels for various diseases based on an individual’s SNP complement. The science behind these services is controversial.
Because GWAS looks for associations between genes and disease, these studies provide data for other research into causes, rather than answering specific questions themselves. An association between a gene difference and a disease does not necessarily mean there is a cause-and-effect relationship. However, some studies have provided useful information about the genetic causes of diseases. For example, three different studies in 2005 identified a gene for a protein involved in regulating inflammation in the body that is associated with a disease-causing blindness called age-related macular degeneration. This opened up new possibilities for research into the cause of this disease. A large number of genes have been identified to be associated with Crohn’s disease using GWAS, and some of these have suggested new hypothetical mechanisms for the cause of the disease.
Pharmacogenomics
Pharmacogenomics involves evaluating the effectiveness and safety of drugs on the basis of information from an individual's genomic sequence. Personal genome sequence information can be used to prescribe medications that will be most effective and least toxic on the basis of the individual patient’s genotype. Studying changes in gene expression could provide information about the gene transcription profile in the presence of the drug, which can be used as an early indicator of the potential for toxic effects. For example, genes involved in cellular growth and controlled cell death, when disturbed, could lead to the growth of cancerous cells. Genome-wide studies can also help to find new genes involved in drug toxicity. The gene signatures may not be completely accurate, but can be tested further before pathologic symptoms arise.
Metagenomics
Traditionally, microbiology has been taught with the view that microorganisms are best studied under pure culture conditions, which involves isolating a single type of cell and culturing it in the laboratory. Because microorganisms can go through several generations in a matter of hours, their gene expression profiles adapt to the new laboratory environment very quickly. On the other hand, many species resist being cultured in isolation. Most microorganisms do not live as isolated entities, but in microbial communities known as biofilms. For all of these reasons, pure culture is not always the best way to study microorganisms. Metagenomics is the study of the collective genomes of multiple species that grow and interact in an environmental niche. Metagenomics can be used to identify new species more rapidly and to analyze the effect of pollutants on the environment. Metagenomics techniques can now also be applied to communities of higher eukaryotes, such as fish.
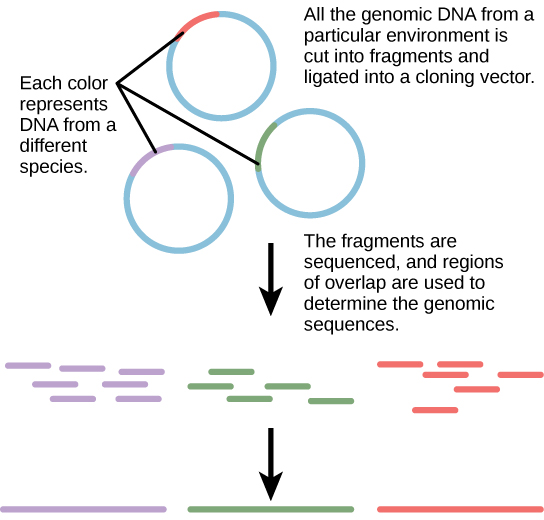
Figure 3. Metagenomics involves isolating DNA from multiple species within an environmental niche. The DNA is cut up and sequenced, allowing entire genome sequences of multiple species to be reconstructed from the sequences of overlapping pieces.
Creation of new biofuels
Knowledge of the genomics of microorganisms is being used to find better ways to harness biofuels from algae and cyanobacteria. The primary sources of fuel today are coal, oil, wood, and other plant products such as ethanol. Although plants are renewable resources, there is still a need to find more alternative renewable sources of energy to meet our population’s energy demands. The microbial world is one of the largest resources for genes that encode new enzymes and produce new organic compounds, and it remains largely untapped. This vast genetic resource holds the potential to provide new sources of biofuels.

Figure 4. Renewable fuels were tested in Navy ships and aircraft at the first Naval Energy Forum. Credit: modification of work by John F. Williams, US Navy
Mitochondrial genomics
Mitochondria are intracellular organelles that contain their own DNA. Mitochondrial DNA mutates at a rapid rate and is often used to study evolutionary relationships. Another feature that makes studying the mitochondrial genome interesting is that in most multicellular organisms, the mitochondrial DNA is passed on from the mother during the process of fertilization. For this reason, mitochondrial genomics is often used to trace genealogy.
Genomics in forensic analysis
Information and clues obtained from DNA samples found at crime scenes have been used as evidence in court cases, and genetic markers have been used in forensic analysis. Genomic analysis has also become useful in this field. In 2001, the first use of genomics in forensics was published. It was a collaborative effort between academic research institutions and the FBI to solve the mysterious cases of anthrax that was transported by the US Postal Service. Anthrax bacteria were made into an infectious powder and mailed to news media and two U.S. Senators. The powder infected the administrative staff and postal workers who opened or handled the letters. Five people died, and 17 were sickened from the bacteria. Using microbial genomics, researchers determined that a specific strain of anthrax was used in all the mailings; eventually, the source was traced to a scientist at a national biodefense laboratory in Maryland.
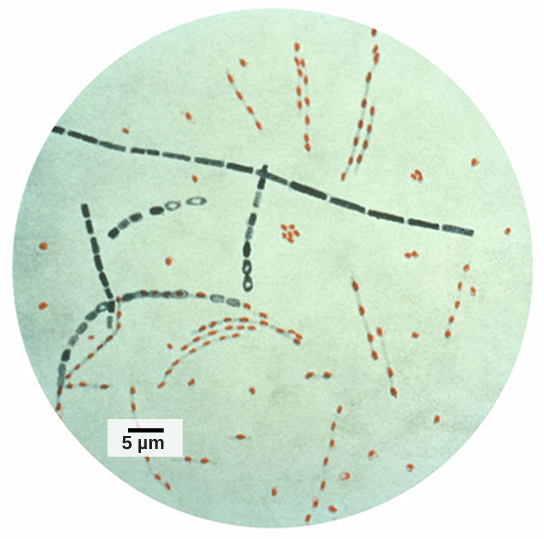
Figure 5. Bacillus anthracis is the organism that causes anthrax. Credit: modification of work by CDC; scale-bar data from Matt Russell
Genomics in agriculture
Genomics can reduce the trials and failures involved in scientific research to a certain extent, which could improve the quality and quantity of crop yields in agriculture. Linking traits to genes or gene signatures helps to improve crop breeding to generate hybrids with the most desirable qualities. Scientists use genomic data to identify desirable traits, and then transfer those traits to a different organism to create a new genetically modified organism, as described in the previous module. Scientists are discovering how genomics can improve the quality and quantity of agricultural production. For example, scientists could use desirable traits to create a useful product or enhance an existing product, such as making a drought-sensitive crop more tolerant of the dry season.

Figure 6. Transgenic agricultural plants can be made to resist disease. These transgenic plums are resistant to the plum pox virus. Credit: Scott Bauer, USDA ARS
Proteomics
Proteins are the final products of genes that perform the function encoded by the gene. Proteins are composed of amino acids and play important roles in the cell. All enzymes (except ribozymes) are proteins and act as catalysts that affect the rate of reactions. Proteins are also regulatory molecules, and some are hormones. Transport proteins, such as hemoglobin, help transport oxygen to various organs. Antibodies that defend against foreign particles are also proteins. In the diseased state, protein function can be impaired because of changes at the genetic level or because of direct impact on a specific protein.
A proteome is the entire set of proteins produced by a cell type. Proteomes can be studied using the knowledge of genomes because genes code for mRNAs, and the mRNAs encode proteins. The study of the function of proteomes is called proteomics. Proteomics complements genomics and is useful when scientists want to test their hypotheses that were based on genes. Even though all cells in a multicellular organism have the same set of genes, the set of proteins produced in different tissues is different and dependent on gene expression. Thus, the genome is constant, but the proteome varies and is dynamic within an organism. In addition, RNAs can be alternatively spliced (cut and pasted to create novel combinations and novel proteins), and many proteins are modified after translation. Although the genome provides a blueprint, the final architecture depends on several factors that can change the progression of events that generate the proteome.
Genomes and proteomes of patients suffering from specific diseases are being studied to understand the genetic basis of the disease. The most prominent disease being studied with proteomic approaches is cancer (Figure 7). Proteomic approaches are being used to improve the screening and early detection of cancer; this is achieved by identifying proteins whose expression is affected by the disease process. An individual protein is called a biomarker, whereas a set of proteins with altered expression levels is called a protein signature. For a biomarker or protein signature to be useful as a candidate for early screening and detection of a cancer, it must be secreted in bodily fluids such as sweat, blood, or urine, so that large-scale screenings can be performed in a noninvasive fashion.
The current problem with using biomarkers for the early detection of cancer is the high rate of false-negative results. A false-negative result is a negative test result that should have been positive. In other words, many cases of cancer go undetected, which makes biomarkers unreliable. Some examples of protein biomarkers used in cancer detection are CA-125 for ovarian cancer and PSA for prostate cancer. Protein signatures may be more reliable than biomarkers to detect cancer cells. Proteomics is also being used to develop individualized treatment plans, which involves the prediction of whether or not an individual will respond to specific drugs and the side effects that the individual may have. Proteomics is also being used to predict the possibility of disease recurrence.

Figure 7. This machine is preparing to do a proteomic pattern analysis to identify specific cancers so that an accurate cancer prognosis can be made. Credit: Dorie Hightower, NCI, NIH
The National Cancer Institute has developed programs to improve the detection and treatment of cancer. The Clinical Proteomic Technologies for Cancer and the Early Detection Research Network are efforts to identify protein signatures specific to different types of cancers. The Biomedical Proteomics Program is designed to identify protein signatures and design effective therapies for cancer patients.
Section summary
Genome mapping is similar to solving a big, complicated puzzle with pieces of information coming from laboratories all over the world. Genetic maps provide an outline for the location of genes within a genome, and they estimate the distance between genes and genetic markers on the basis of the recombination frequency during meiosis. Physical maps provide detailed information about the physical distance between the genes. The most detailed information is available through sequence mapping. Information from all mapping and sequencing sources is combined to study an entire genome.
Whole genome sequencing is the latest available resource to treat genetic diseases. Some doctors are using whole genome sequencing to save lives. Genomics has many industrial applications including biofuel development, agriculture, pharmaceuticals, and pollution control.
Imagination is the only barrier to the applicability of genomics. Genomics is being applied to most fields of biology; it can be used for personalized medicine, prediction of disease risks at an individual level, the study of drug interactions before the conduction of clinical trials, and the study of microorganisms in the environment as opposed to the laboratory. It is also being applied to the generation of new biofuels, genealogical assessment using mitochondria, advances in forensic science, and improvements in agriculture.
Proteomics is the study of the entire set of proteins expressed by a given type of cell under certain environmental conditions. In a multicellular organism, different cell types will have different proteomes, and these will vary with changes in the environment. Unlike a genome, a proteome is dynamic and under constant flux, which makes it more complicated and more useful than the knowledge of genomes alone.