
21.1: Sickle Cell Anemia
- Page ID
- 8539
Sickle cell anemia: a look at the connection between DNA and phenotype
Genes are translated into proteins; mutations often (but not always) result in changes in the sequence of amino acids in those proteins. Changes in the amino acid sequence can modify (in various ways) or even completely destroy protein function. Proteins have many functions within cells, and a change in those functions results in a change in the phenotype of that cell or organism. So a mutation as simple as a single base change in a DNA sequence can have dramatic effects on phenotype. One of the best examples of this phenomenon can be observed when mutations occur in the gene for one of the protein components of the red blood cell protein we call hemoglobin.
A major component of the erythrocytes (red blood cells) found in vertebrates is hemoglobin. A molecule of hemoglobin from a normal adult human contains four proteins (two identical alpha polypeptides and two identical beta polypeptides) surrounding a core of heme (complex molecule containing an atom of iron which can combine reversibly with oxygen). Thus, hemoglobin functions as the major oxygen-carrying constituent of blood. Because of hemoglobin, a given volume of blood can carry far more oxygen than could be dissolved in an equal volume of water.
In many human populations, particularly those with origins in Central Africa or the Mediterranean, there are individuals who suffer from severe anemia and whose blood contains numerous distorted, sickle-shaped erythrocytes. Hence, the disease was given the name sickle cell anemia.
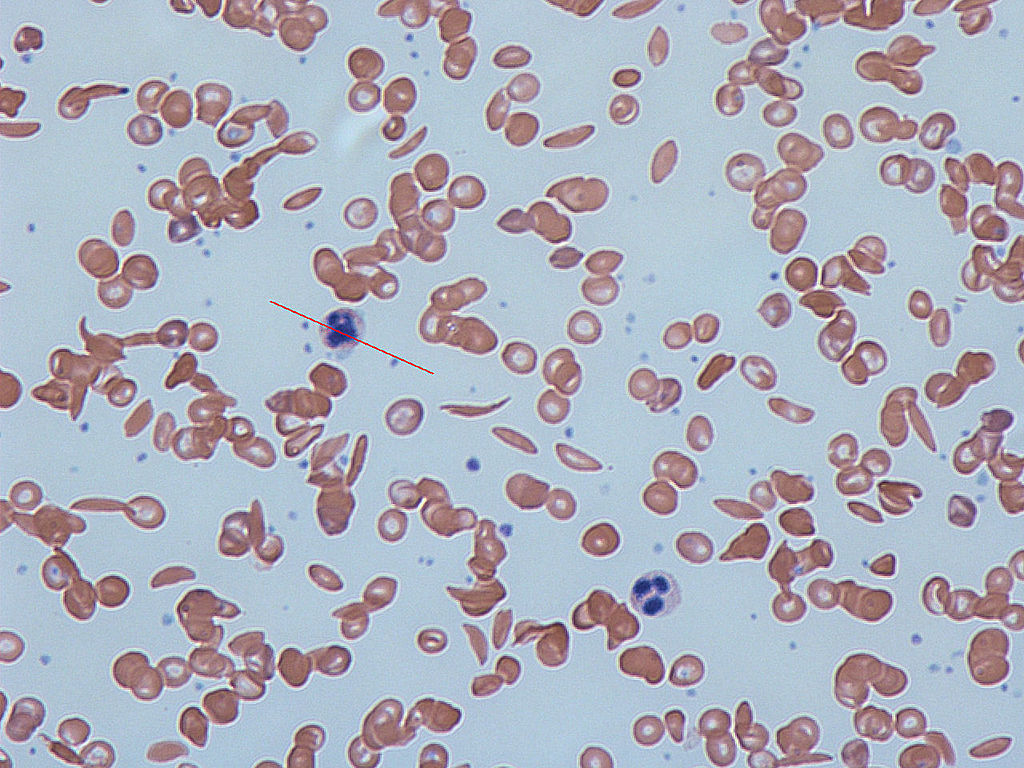
Figure 1. Notice the sickle shaped cells in the image by Dr Graham Beards via Wikimedia Commons
Biochemical studies established that the gene affected in sickle-cell anemia has the code for an abnormal beta polypeptide, which is one of the components of the hemoglobin molecule. Therefore, there are two different forms of the hemoglobin gene that codes for the beta chain:
- Form 1: HbA has the code for a normal beta chain
- Form 2: HbS has the code for an abnormal beta chain
Humans are diploid organisms; they have two copies of most genes. However, the two copies they possess do not have to be identical. When there are two possible alleles for a gene (such as in the gene for the beta chain of hemoglobin), a diploid individual will have one of three possible combinations of the two alleles. They can be HbA HbA , HbA HbS, or HbSHbS.
The set of alleles present in an individual for a given gene is known as the individual’s genotype. The three combinations of two alleles above are therefore the three different genotypes. Individuals that have two copies of the same allele are called homozygous; individuals with two different alleles are called heterozygous. So an individual that is HbA HbA is homozygous normal beta chain; an individual that is HbA HbS is heterozygous; and an individual that is HbS HbS is homozygous abnormal beta chain. It is the homozygous HbS individuals that contain sickle-shaped blood cells.
Mechanism of the disease
In the capillaries (microscopic blood vessels that directly exchange oxygen with the tissues), erythrocytes can be subjected to low oxygen tension after they lose their oxygen to the surrounding tissues. In this low oxygen situation, the abnormal hemoglobin molecules of HbS HbS individuals tend to polymerize (join together), forming stiff, tubular fibers which ultimately distort the shape of the entire erythrocyte, giving it the characteristic “sickle” shape. These sickled cells have a number of effects on the body via two processes.
- Sickled cells are less able to enter and move through the capillaries: Once in the capillaries, they clog capillary flow and cause small blood clots. Reduced blood flow results in reduced oxygen availability to the tissues. Reduced oxygen supply results in tissue death and damage to vital organs (e.g., the heart, liver and spleen).
- Sickled blood cells have a shorter lifespan than normal red blood cells: Reduced lifespan of erythrocytes places a greater demand on the bone marrow to make new red blood cells and on the spleen to break down dead erythrocytes. Increased demand on the bone marrow results in severe pain in the long bones and joints. Individuals suffering from sickle cell anemia are frequently ill and generally have a considerably reduced lifespan. These individuals are said to have sickle cell disease.
Heterozygous individuals (HbA HbS) are said to be carriers for sickle cell anemia. Note that this is a specific term and is not the same thing as sickle cell anemia—heterozygotes do not have the disease themselves but their children may inherit the condition. Carriers have no anemia, have good health (as do HbA HbA individuals), and their erythrocytes maintain normal shape in the blood. In other words, they are phenotypically normal under most conditions, and probably do not know that they “carry” the HbS allele. However, if heterozygotes are exposed to conditions of low oxygen levels (such as strenuous activity at high altitudes), some of their erythrocytes do sickle. Red blood cells in blood samples of heterozygotes subjected to greatly reduced oxygen tension in the laboratory also sickle.
Why is sickle cell anemia most prevalent in people with origins in Central Africa and the Mediterranean? If you look at Figure 2, you will see the occurrence of sickle cell anemia overlaps with the pervasiveness of malaria. This seems odd, but those individuals who are heterozygous (HbA HbS) for the sickle cell allele are less likely to contract and die from malaria then those who are homozygous (HbA HbA). The HbS polypeptide that is produced by the heterozygous individual stops the organism (Plasmodium) that causes malaria from invading the red blood cells. So, in areas where malaria is common there is selection pressure for the HbS allele, and the HbS allele occurs in a higher frequency because the those who have one copy of the HbS allele will live longer and have more children. In areas where malaria is not common, there is selection pressure against the HbS allele, and the HbS allele occurs in a lower frequency. As you will learn in a later chapter, there is an 25% chance that two carriers will have a child who is homozygous HbS HbS), and this child will pay the evolutionary price for the protection from malaria that the parents were afforded. It seems to be in this way that evolution by natural selection retains such a potentially detrimental allele in a population. The sickle cell example is only one of what is called heterozygous advantage; we have provided a number of other examples in Table 1.
|
Distribution of malaria and the frequency of sickle cell allele
|
|---|

| Recessive illness | Heterozygote advantage | Possible explanation |
|---|---|---|
| Cystic fibrosis | Protection against diarrheal diseases such as cholera | Carriers have too few functional chloride channels in intestinal cells, blocking toxin |
| G6PD Deficiency | Protection against malaria | Red blood cells inhospitable to malaria |
| Phenylketonuria (PKU) | Protection against miscarriage induced by a fungal toxin | Excess amino acid (phenylalanine) in carriers inactivates toxin |
| Tay-Sachs disease | Protection against tuberculosis | Unknown |
| Noninsulin-dependent diabetes mellitus | Protection against starvation | Tendency to gain weight protects against starvation during famine |