
1.17: Protein Structure
- Page ID
- 8610
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Amino Acid Structure
Amino acids are the monomers that make up proteins. Each amino acid has the same core structure, which consists of a central carbon atom, also known as the alpha (α) carbon, bonded to an amino group (NH2), a carboxyl group (COOH), and a hydrogen atom. Every amino acid also has another atom or group of atoms bonded to the alpha carbon known alternately as the R group, the variable group or the side-chain. For an additional introduction on amino acids, click here for a short (4 minute) video.
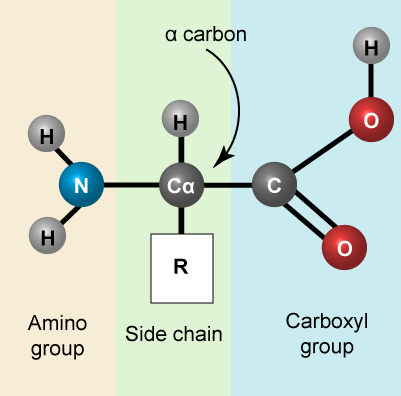
Amino acids have a central asymmetric carbon to which an amino group, a carboxyl group, a hydrogen atom, and a side chain (R group) are attached. Attribution: Marc T. Facciotti (own work)
Possible discussion:
Recall that one of the learning goals for this class is that you (a) be able to recognize, in a molecular diagram, the backbone of an amino acid and its side chain (R-group) and (b) that you be able to draw a generic amino acid. Make sure that you practice both. You should be able to recreate something like the figure above from memory (a good use of your sketchbook is to practice drawing this structure until you can do it with the crutch of a book or the internet).
The protein backbone
The name "amino acid" is derived from the fact that all free amino acids contain both an amino group and carboxylic acid group. These will be used to make the peptide bonds between amino acids in a protein (only the amino groups at very beginning (the "N terminus") and the carboxyl group at the very end (the C terminus) will remain in a polypeptide (= protein). There are 20 genetically encoded amino acids available to the cell to build in proteins and all of these contain the same core sequence:
N-C-C-
where the first ("alpha") C will always carry the R group and the second will have a double (ketone) bond to oxygen. The amino acids are arranged in a single line- there are no branches. When looking at a chain of amino acids it is always helpful to first orient yourself by finding this backbone pattern starting from the N terminus to the C terminus. When we write the sequence of a protein, we will always write it from the from "N to C".

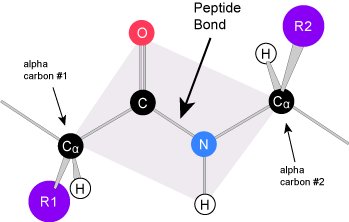
Peptide bond formation is a condensation reaction. The carboxyl group of the first amino acid is linked to the amino group of the second incoming amino acid. In the process, a molecule of water is released and a peptide bond is formed. Try finding the backbone in the dipeptide formed from this reaction. The pattern you are looking for is: N-C-C-N-C-C
The sequence and the number of amino acids ultimately determine the protein's shape, size, and function. Each amino acid is attached to another amino acid by a covalent bond, known as a peptide bond, which is formed by a dehydration synthesis (= condensation) reaction. The carboxyl group of one amino acid and the amino group of the incoming amino acid combine, releasing a molecule of water and creating the peptide bond.
Amino Acid R group
The amino acid R group is a term that refers to the variable group on each amino acid. The amino acid backbone is identical on all amino acids (though proline is a bit odd- check it out), and the R groups are different on all amino acids. For the structure of each amino acid refer to the figure below.

There are 20 common amino acids found in proteins, each with a different R group (variant group) that determines its chemical nature. R-groups are circled in teal. Charges are assigned assuming pH ~6.0. The full name, three letter abbreviation and single letter abbreviations are all shown. Attribution: Marc T. Facciotti (own work)
Note:
Possible Discussion: Let's think about the relevance of having 20 different amino acids. If you were using biology to build proteins from scratch, how might it be useful if you had 10 more additional amino acids at your disposal? By the way, this is actually happening in a variety of research labs - why would this be potentially useful?
Each variable group on an amino acid gives that amino acid specific chemical properties (acidic, basic, polar, or nonpolar). This gives each amino acid R group different chemical properties.
For example, amino acids such as valine, methionine, and alanine are typically nonpolar or hydrophobic in nature, while amino acids such as serine and threonine have polar character and possess hydrophilic side chains.
Note:
Practice: Using your knowledge of functional groups, try classifying each amino acid in the figure above as either having the tendency to be polar or nonpolar. Try to find other classification schemes and think make lists for yourself of the amino acids you would put into each group. You can also search the internet for amino acid classification schemes - you will notice that there are different ways of grouping these chemicals based on chemical properties. You may even find that there are contradictory schemes. Try to think about why this might be and apply your chemical logic to figuring out why certain classification schemes were adopted and why specific amino acids were placed in certain groups.
Protein Folding and Structure
To understand how the protein gets its final shape or conformation, we need to understand the four levels of protein structure: primary, secondary, tertiary, and quaternary. For a short (4 minutes) introduction video on protein structure click here.
Primary Structure
The unique sequence of amino acids in a polypeptide chain is its primary structure. The linear sequence of amino acids in the polypeptide chain are held together by peptide bonds and result in the N-C-C-N-C-C patterned backbone. The primary structure is coded for in the DNA, a process you will learn about in the Transcription and Translation modules.
Secondary structure
The local folding of the polypeptide in some regions gives rise to the secondary structure of the protein. The most common shapes created by secondary folding are the α-helix and β-pleated sheet structures. These secondary structures are held together by hydrogen bonds forming between the backbones of amino acids in close proximity to one another. More specifically, the oxygen atom in the carboxyl group from one amino acid can form a hydrogen bond with a hydrogen atom bound to the nitrogen in the amino group of another amino acid. In the alpha helix, this partnering amino acid is always four amino acids farther along the chain. The hydrogen bonding in alpha helices stabilizes the formation of a rigid cylinder of amino acids. In a beta pleated sheet (shown below) the hydrogen bonded partners might be very far away from each other in the primary structure of the protein (i.e., the 15th and 100th amino acids in the chain) but the secondary structure holds these amino acids in close proximity to one another.
Practice: Note that in the structure of the beta pleated sheet drawn below one chain is drawn in the N to C direction while the other is drawn C to N (stop to try to figure out what I mean by this, and which strand is drawn which way- this is great practice). Beta pleated sheets can be formed both through either the antiparallel or the parallel alignment of the peptide chains, though the former structures are more stable.

Tertiary Structure
The unique three-dimensional structure of a polypeptide is its tertiary structure. This structure is in part due to chemical interactions at work on the polypeptide chain. Primarily, the interactions among R groups creates the complex three-dimensional tertiary structure of a protein. The nature of the R groups found in the amino acids involved can counteract the formation of the hydrogen bonds described for standard secondary structures. For example, R groups with like charges are repelled by each other and those with unlike charges are attracted to each other (ionic bonds). When protein folding takes place (in an aqueous compartment), the hydrophobic R groups of nonpolar amino acids will cluster together in the interior of the protein, whereas the hydrophilic R groups lay on the outside. These types of interactions are known as hydrophobic interactions. Interaction between cysteine side chains can form disulfide linkages in the presence of oxygen, this is the only covalent bond that specifically stabilizes tertiary structure. As you'll recall, covalent bonds are about 10x stronger that hydrogen or ionic bonds in an aqueous environment. Thus disufide bonds can make a protein's tertiary structure more resistant to denaturing influences like heat or salt.

All of these interactions, weak and strong, determine the final three-dimensional shape of the protein. When a protein loses its three-dimensional shape, it may no longer be functional. However, some small proteins, with established disulfide bonds, can refold even after boiling. This is because the disulfide bonds (established during the initial folding of the protein, during its synthesis), reduce the number of possible ways to "misfold".
Quaternary Structure
In nature, some proteins are formed from multiple proteins, also known as subunits, and the interaction of these subunits forms the quaternary structure. Weak interactions between the subunits help to stabilize the overall structure. For example, hemoglobin consists of two subunits, encoded by the alpha- and betaglobin genes. This protein complex also carries the prosthetic group hemoglobin. The multi-subunit structure of this protein complex gives it regulatory characteristics not shared by its single-subunit cousin, myoglobin.

Denaturation and Protein Folding
Given that the role of a proteins is to assume a certain shape that will facilitate a certain process, all levels of protein structure (primary through quaternary) are critical to protein function. We understand how primary structure is established (the linear array of amino acids is encoded in the linear array of bases in DNA- the rest is a matter of transcription and translation). We also know how secondary structures self-assemble, and can predict these structures with a certain amount of confidence, based on the primary sequence of amino acids. However, our understanding of the establishment of the tertiary structure is still a work in progress. Think about this- the protein can twist between each amino acid and there can be hundreds of amino acids in a protein, resulting in an enormous number of possible shapes, even given the relative rigidity of the alpha helices and beta sheets. It was originally thought that the proteins themselves were responsible for the folding process; that they would simply fold into the lowest potential energy structure (although finding this structure via random folding might take a ridiculously long time (several billion years), and the protein might be "trapped" in semi-stable but incorrect structures). However, we have since found that many proteins receive assistance in the folding process from protein helpers known as chaperones (or chaperonins) that associate with the target protein during the process of translation and folding. They act by preventing random aggregation of the amino acid chains that make up the complete protein structure, thus limiting the number of choices available for random folding. They disassociate from the protein once it has folded.
Each protein has its own unique sequence and shape that are held together by chemical interactions. If the protein is subject to changes in temperature, pH, or exposure to chemicals, the protein structure may change, losing its shape without losing its primary sequence. This process is known as denaturation. Denaturation is sometimes reversible because the primary structure of the polypeptide is conserved in the process- only higher order structures are lost. A very short protein or, as mentioned above, a protein stabilized by many covalent disulfide bonds might be able to refold effectively and regain its function. Generally, however, denaturation is irreversible, leading to permanent loss of function. One example of irreversible protein denaturation is when an egg is boiled. The albumin protein in the liquid egg white unravels when it heats up, as the hydrogen and ionic bonds that stabilize its higher order structure fall apart. When cooled, these interactions resume, but between different partners, resulting in stable but incorrect tertiary structures. Not all proteins are denatured at high temperatures; for instance, bacteria that survive in hot springs have proteins that function at temperatures close to boiling. The stomach is also very acidic, has a low pH, and it denatures proteins as part of the digestion process; however, the digestive enzymes of the stomach retain their activity under these conditions.
Question:
How might these proteins (in extremeophile bacteria, and in the stomach) stabilize their higher-order structure?