
14: Gel Electrophoresis
- Page ID
- 141653
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Summary
Gel electrophoresis is a method that separates biological macromolecules based on their size and sometimes (if a non-denaturing gel is used) their 3D structure.
Also known as
Variants: DNA/RNA gel, protein gel, agarose gel, polyacrylamide gel, SDS-PAGE
Samples needed
Mixtures or pure samples of either nucleic acids or proteins
Method
To separate a mixture of biological macromolecules, gel electrophoresis takes advantage of the sieving effects of a polymer gel. Generally, an electric field is applied so that negatively charged molecules travel toward the bottom of the gel. Smaller molecules move more quickly, as they pass through the crosslinked polymer more readily than large molecules, so small molecules end up near the bottom of the gel. (Note that nucleic acids are inherently negatively charged due to the phosphate backbone, but proteins need an appropriate buffer to be coated in negative charge.)
Gel electrophoresis is most frequently used as an analytical method, i.e. to determine the size and number of molecules in a sample. However, sometimes specific bands are cut out of the gel and purified for further use.
Gel electrophoresis can be used to separate samples containing nucleic acids and/or proteins. When viewing an image of a gel, it is important to determine which type of molecules are being visualized. After being run, gels are normally clear and colorless. Therefore, further steps are necessary to visualize the bands in the gel. For nucleic acids, a common method is the use of intercalating agents like ethidium bromide, which is visualized under a UV light. For proteins, a common method is using Coomassie brilliant blue dye, which binds to proteins relatively non-specifically.
The two most common types of gels used in electrophoresis are made of either agarose or polyacrylamide. Agarose is mainly used for separating nucleic acids with relatively large size differences of at least a few 100 nucleotides. Polyacrylamide gives greater resolution and is normally used for proteins and for separating nucleic acids with much smaller size differences. For instance, polyacrylamide gels are used for sequencing gels, where nucleic acids that differ by only one nucleotide need to be separated. Both types of gels can be made using different percentages of the polymer to achieve different separation characteristics.
Gel electrophoresis can be run under denaturing or non-denaturing (native) conditions, depending on whether the researcher wants to maintain secondary and higher structures of the macromolecules and interactions between macromolecules. For instance, SDS-PAGE is a very common technique used to separate denatured proteins, so that the proteins run through the gel based on only their molecular weight and not their structures. SDS-PAGE stands for sodium dodecyl sulfate polyacrylamide gel electrophoresis. SDS is an ionic detergent, which along with a reducing agent like Dithiothreitol (DTT) or β-mercaptoethanol (BME), and a heating step, ensures that proteins are fully denatured before running them on the gel. SDS also coats proteins in negative charge proportional to their molecular weight, so that they move toward the positive electrode at the bottom of the gel.
When imaging nucleic acids on a gel, supercoiling also has to be taken into account. Most commercially sold molecular weight ladders for nucleic acids consist of linear molecules, so a piece of linear nucleic acid will run “true to size” in comparison with the ladder. However, a nicked, circular, double-stranded nucleic acid will run slower than expected. Conversely, a supercoiled, circular, double-stranded nucleic acid will run more quickly than expected. This is true regardless of whether the supercoiling is positive or negative.
Gel electrophoresis is often used as an early step in other methods that detect one specific macromolecule, like Southern blot
Controls
Molecular weight markers made of macromolecules of known size should be run alongside of samples to relate distance traveled in the gel to molecular weight of bands.
Interpretation
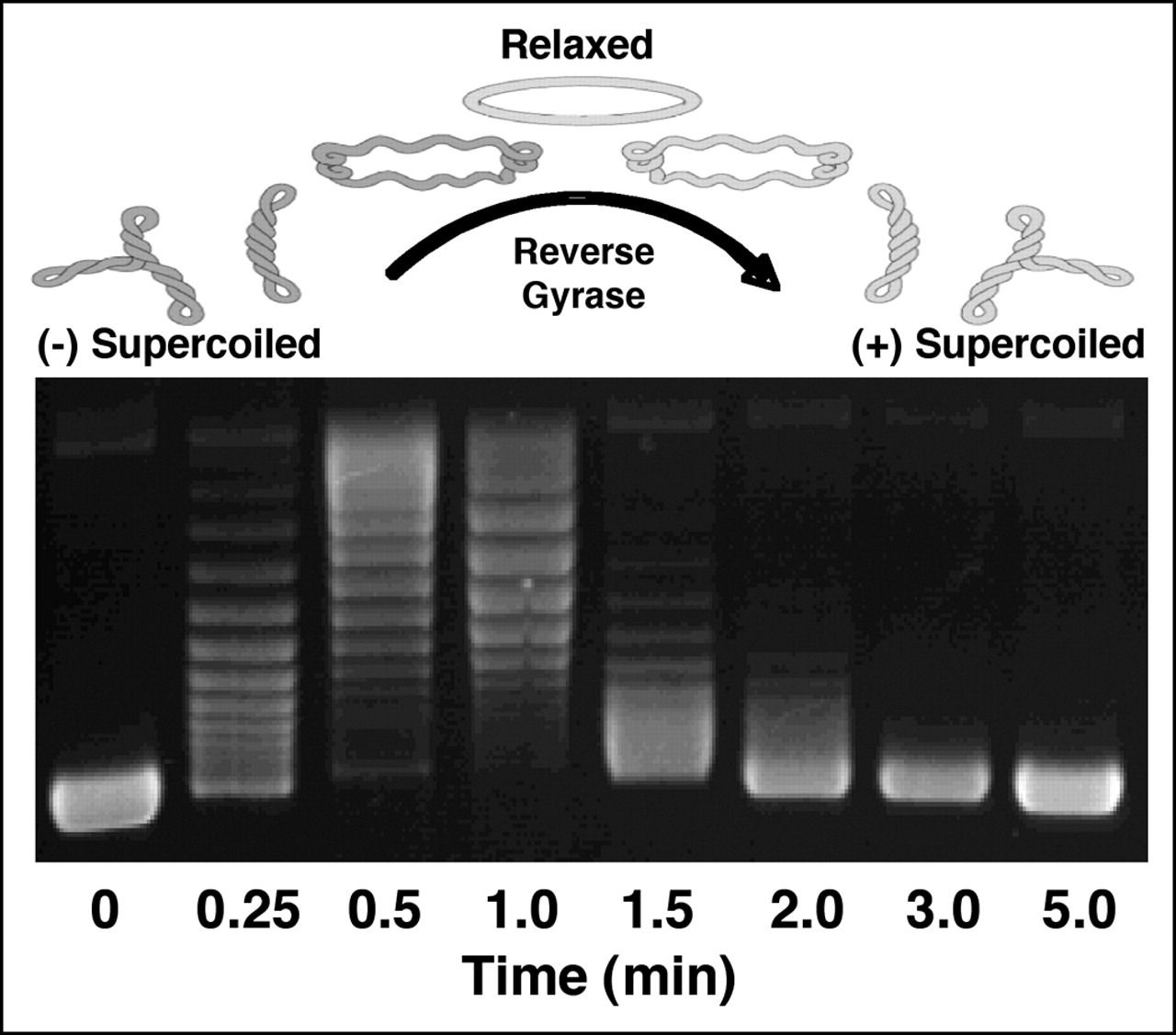 Figure 1. Ethidium bromide stained agarose gel loaded with plasmid (circular dsDNA). Relevant section of caption for published figure reads: “Figure 2. Generation of positively supercoiled DNA by A. fulgidus reverse gyrase. The positions of negatively (-) supercoiled, relaxed, and positively (+) supercoiled DNA are indicated. Negatively supercoiled pBR322 plasmid DNA was incubated with reverse gyrase for the indicated times. The extent of positive supercoiling was monitored by agarose gel electrophoresis.” “Figure 2” by McClendon et al.[1] [Image description]
Figure 1. Ethidium bromide stained agarose gel loaded with plasmid (circular dsDNA). Relevant section of caption for published figure reads: “Figure 2. Generation of positively supercoiled DNA by A. fulgidus reverse gyrase. The positions of negatively (-) supercoiled, relaxed, and positively (+) supercoiled DNA are indicated. Negatively supercoiled pBR322 plasmid DNA was incubated with reverse gyrase for the indicated times. The extent of positive supercoiling was monitored by agarose gel electrophoresis.” “Figure 2” by McClendon et al.[1] [Image description]In this experiment, researchers purified a circular, dsDNA molecule (plasmid) called pBR322 from E. coli. In vivo, the vast majority of DNA is negatively supercoiled, so unless the plasmid has an unusual secondary structure, it is likely negatively supercoiled when purified from the bacterial cells. Aliquots of the plasmid were then treated with purified enzyme, reverse gyrase, from the microbe A. fulgidus for varying amounts of time. When the plasmid is treated for a short time with reverse gyrase, a series of bands further up on the gel (compared to the 0 min treatment) appear. These correspond to the plasmid becoming less supercoiled, in this case, less negatively supercoiled. Each band is a different topoisomer, i.e. the same plasmid with a different number of supercoils. As the times get longer, the bands start to move back down the gel, indicating an increase in supercoiling. In the absence of evidence to the contrary, we assume that the DNA started out as negatively supercoiled. Therefore, the conclusion is that reverse gyrase increases positive supercoiling, initially removing negative supercoils, and ultimately adding positive supercoils.
Image Descriptions
Figure 1 image description:
A DNA gel. Lanes indicate the amount of time the sample is treated with reverse gyrase. At t = 0 min, there is one band near the bottom of the gel. At t = 0.25 min, there are a series of bands further up the gel. The series of bands moves up the gel & then back down the gel until by t = 3 min nearly all the DNA is present in one band at the same position as the band present at t = 0 min. ↵
Thumbnail
"Gel electrophoresis 2.jpg"↗ by Mnolf is licensed under CC BY-SA 3.0↗.
Description: Gel electrophoresis of DNA made visible using ethidium bromide and ultraviolet light.
Author
Katherine Mattaini, Tufts University
-
McClendon, A. K., A. C. Rodriguez, and N. Osheroff. 2005. Human Topoisomerase IIα Rapidly Relaxes Positively Supercoiled DNA: IMPLICATIONS FOR ENZYME ACTION AHEAD OF REPLICATION FORKS*. Journal of Biological Chemistry 280:39337–39345. ↵