7.4: DNA Repair
- Page ID
- 7845

Source: BiochemFFA_7_3.pdf. The entire textbook is available for free from the authors at http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy

Safeguarding the genome
In the last section we considered the ways in which cells deal with the challenges associated with replicating their DNA, a vital process for all cells. It is evident that if DNA is the master copy of instructions for an organism, then it is important not to make mistakes when copying the DNA to pass on to new cells. Although proofreading by DNA polymerases greatly increases the accuracy of replication, there are additional mechanisms in cells to further ensure that newly replicated DNA is a faithful copy of the original, and also to repair damage to DNA during the normal life of a cell.
DNA damage
All DNA suffers damage over time, from exposure to ultraviolet and other radiation, as well as from various chemicals in the environment (Figures 7.34 & 7.35). Even chemical reactions naturally occurring within cells can give rise to compounds that can damage DNA. As you already know, even minor changes in DNA sequence, such as point mutations can sometimes have far-reaching consequences. Likewise, unrepaired damage caused by radiation, environmental chemicals or even normal cellular chemistry can interfere with the accurate transmission of information in DNA. Maintaining the integrity of the cell's "blueprint" is of vital importance and this is reflected in the numerous mechanisms that exist to repair mistakes and damage in DNA.
Post-replicative mismatch repair
We earlier discussed proofreading by DNA polymerases during replication. While proofreading significantly reduces the error rate, not all mistakes are fixed on the fly by DNA polymerases.
What mechanisms exist to correct the replication errors that are missed by the proof-reading function of DNA polymerases? Errors that slip by proofreading during replication can be corrected by a mechanism called mismatch repair. While the error rate of DNA replication is about one in 107 nucleotides in the absence of mismatch repair, this is further reduced a hundred-fold to one in 109 nucleotides when mismatch repair is functional.
What are the tasks that a mismatch repair system faces?
It must:
- Scan newly made DNA to see if there are any mispaired bases (e.g., a G paired to a T)
- Identify and remove the region of the mismatch.
- Correctly fill in the gap created by the excision of the mismatch region.
Distinguishing strands
Importantly, the mismatch repair system must have a means to distinguish the newly made DNA strand from the template strand, if replication errors are to be fixed correctly. In other words, when the mismatch repair system encounters an A-G mispair, for example, it must know whether the A should be removed and replaced with a C or if the G should be removed and replaced with a T.
But how does the mismatch repair system distinguish between the original and the new strands of DNA? In bacteria, the existence of a system that methylates the DNA at GATC sequences is the solution to this problem. E.coli has an enzyme, DNA adenine methylase (Dam) that adds methyl groups on the to adenines in GATC sequences in DNA (Figure 7.36). Newly replicated DNA has not yet undergone methylation and thus, can be distinguished from the template strand, which is methylated.
The mismatch repair proteins selectively replace the strand lacking methylation, thus ensuring that it is mistakes in the newly made strand that are removed and replaced. Because methylation is the criterion that enables the mismatch repair system to choose the strand that is repaired, the bacterial mismatch repair system is described as being methyl-directed.
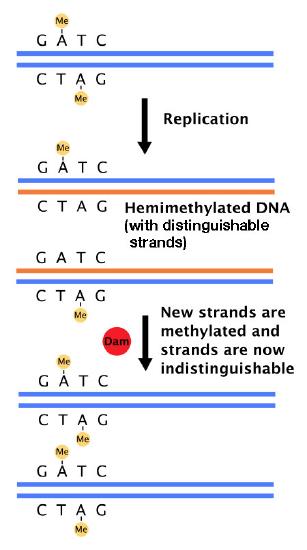
Figure 7.36- Dam methylase adds methyl groups at GATC sequences
Mismatch repair genes
Mismatch repair has been well studied in bacteria, and the proteins involved have been identified. In E.coli, mismatch repair proteins are encoded by a group of genes collectively known as the mut genes. Important components of the mismatch repair machinery are the proteins MutS, MutL and MutH (Figure 7.37).
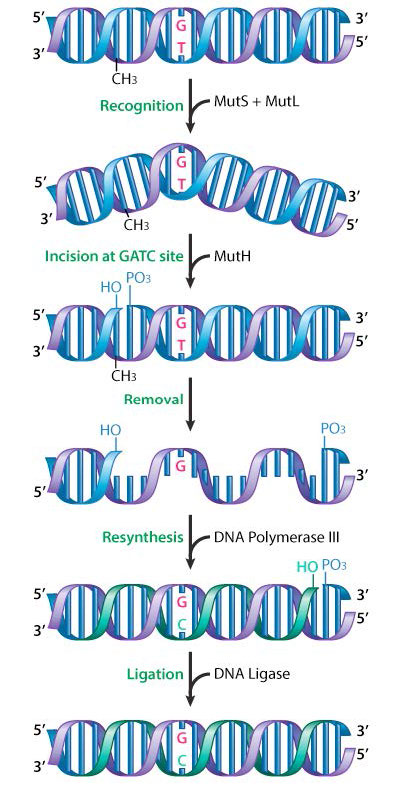
MutS acts to recognize the mismatch, while MutL and MutH are recruited to the mismatch site by the binding of MutS. MutH is an endonuclease that cuts the newly synthesized and, as yet, unmethylated DNA strand at a GATC. This activates a DNA helicase and an exonuclease that help unwind and remove the region containing the mismatch. DNA polymerase III fills in the gap, using the opposite strand as the template, and ligase joins the ends, to restore a continuous strand.
Eukaryotes also have a mismatch repair system that repairs not only single base mismatches but also insertions and deletions. Homologs to the E. coli MutS and MutL have been identified in other organisms, including humans: hMSH1 and hMSH2 (human MutS homolog 1 and 2) are homologous to MutS, while hMLH 1 is homologous to MutL. These, together with additional proteins, carry out mismatch repair in eukaryotic cells.
DNA methylation is not used by eukaryotic cells as a way to distinguish the new strand from the template, and it is not yet completely understood how the mismatch repair system in eukaryotes "knows" which strand to repair. There is evidence that the newly made DNA may be recognized by the fact that it is nicked, or discontinuous. This suggests that discontinuity resulting from Okazaki fragments that have not yet been joined together may permit the new strand to be distinguished from the old, continuous template strand.
Repairing damage to DNA
In the preceding section we looked at mistakes made when DNA is copied, where the wrong base is inserted during synthesis of the new strand. But even DNA that is not being replicated can get damaged or mutated. These sorts of damage are not associated with DNA replication, rather they can occur at any time.
What causes damage to DNA?
Some major causes of DNA damage are:
a. Radiation (e.g., UV rays in sunlight and in tanning booths, or ionizing radiation)
b. Exposure to damaging chemicals, such as nitrosamines or polycyclic aromatic hydrocarbons, in the environment (see Figure 7.38)
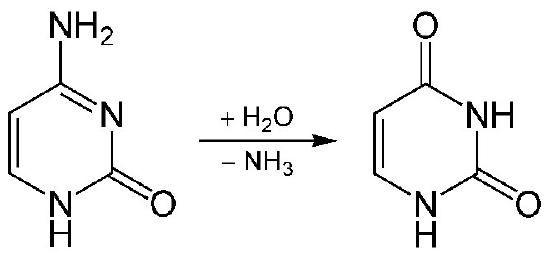
c. Chemical reactions within the cell (such as the deamination of cytosine to give uracil, or the methylation of guanine to produce methylguanine).
This means the DNA in your cells is vulnerable to damage simply from normal sorts of actions, such as walking outdoors, being in traffic, or from the chemical transformations occurring in every cell as part of its everyday activities. (Naturally, the damage is much worse in situations where exposure to radiation or damaging chemicals is greater, such as when people use tanning beds, or smoke, regularly.)
Types of damage
What kinds of damage do these agents cause? Radiation can cause different kinds of damage to DNA.
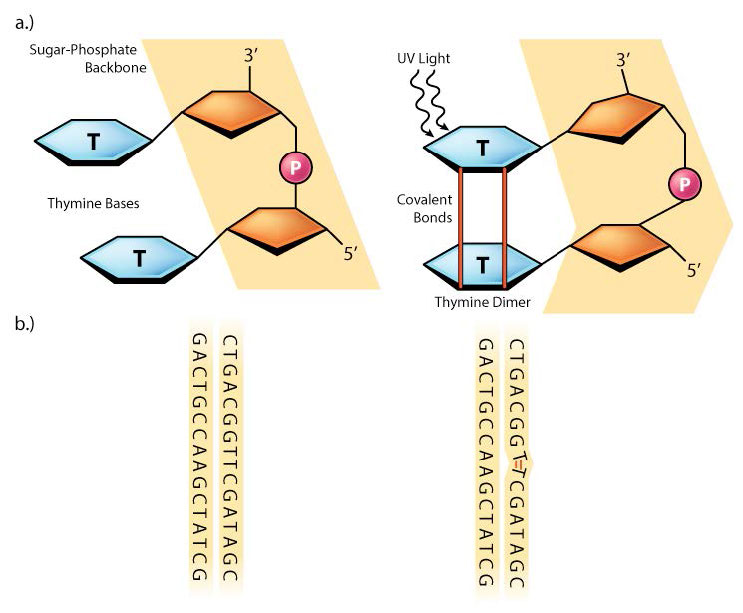
Sometimes, as with much of the damage done by UV rays, two adjacent pyrimidine bases in the DNA will be cross-linked to form cyclobutane pyrimidine dimers or CPDs (see Figure 7.39). Note that these are two neighboring pyrimidine bases on the same strand of DNA. UV exposure can also lead to the formation of another type of lesion, known as a (6-4) photoproduct or 6-4PP (Figure 7.39). Ionizing radiation can cause breaks in the DNA backbone, in one or both strands.
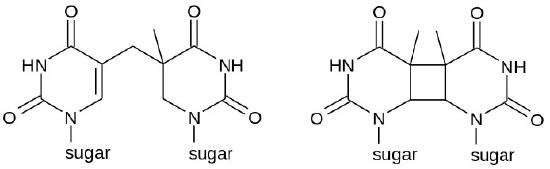
Figure 7.39 - Possible chemical structures of a pyrimidine dimer - 6-4PP (left) and CPD (right) - Wikipedia
Molecules like benzopyrene, found in automobile exhaust, can attach themselves to bases, forming bulky DNA adducts in which large chemical groups are linked to bases in the DNA. Damage like pyrimidine dimers, 6-4PPs or chemical adducts can physically distort the DNA helix, causing DNA and RNA polymerase to stall when they attempt to copy those regions of DNA (Figure 7.40).
Chemical reactions occurring within cells can cause cytosines in DNA to be deaminated to uracil. Other sorts of damage in this category include the formation of oxidized bases like 8-oxo-guanine or alkylated bases like O6-methylguanine. These do not actually change the physical structure of the DNA helix, but they can cause problems because uracil and 8-oxo-guanine pair with different bases than the original cytosine or guanine, leading to mutations on the next round of replication. O6-methylguanine similarly can form base pairs with thymine instead of cytosine.
Removing damage
Cells have several ways to remove the sorts of damage described above. The first of these is described as direct reversal. Many organisms (though, unfortunately for us, not humans) can repair UV damage like CPDs and 6-4PPs because they possess enzymes called photolyases (photo=light; lyase=breakdown enzyme - Figure 7.41). Photolyases work through a process called photoreactivation, and use blue light energy to catalyze a photochemical reaction that breaks the aberrant bonds in the damaged DNA and returns the DNA to its original state.
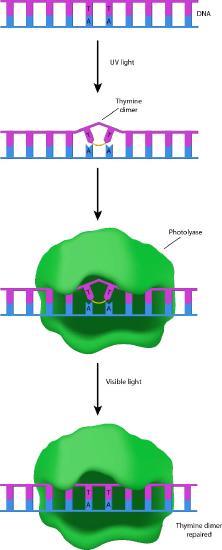
Suicide enzyme
O6-methylguanine in DNA can also be removed by direct reversal, with the help of the enzyme O6-methylguanine methyltransferase. This is a very unusual enzyme that removes the methyl group from the guanine and transfers it onto a cysteine residue in the enzyme. The addition of the methyl group to the cysteine renders the enzyme non-functional.
As you know, most enzymes are catalysts that remain unchanged over the course of the reaction, permitting a single enzyme molecule to repeatedly catalyze a reaction. Because the O6-methylguanine methyltransferase does not fit this description, it is sometimes not regarded as a true enzyme. It has also been called a suicide enzyme, because the enzyme “dies” as a result of its own activity.
Excision repair
Excision repair is another common strategy. Excision repair is a general term for the cutting out and re-synthesizing of the damaged region of a DNA. There are several different kinds of excision repair, but they all involve excising the portion of the DNA that is damaged, followed by repair synthesis using the other strand as template, and finally, ligation to restore continuity to the repaired strand. Cells possess several different kinds of excision repair, each geared to specific kinds of DNA damage. Between them, these repair systems deal with the wide variety of insults to the genome.
Nucleotide excision repair
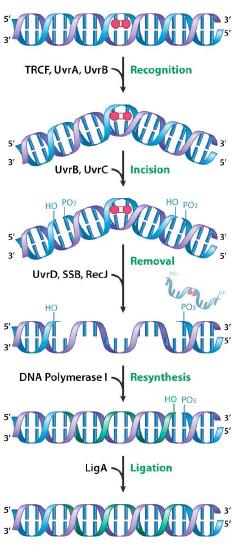
Nucleotide excision repair (NER) fixes damage such as the formation of chemical adducts, as well as UV damage. Both chemical adducts and the formation of CPDs or 6,4 photoproducts can cause significant distortion of the DNA helix. NER proteins act to cut the damaged strand on either side of the lesion. A short portion of the DNA strand containing the damage is then removed and a DNA polymerase fills in the gap with the appropriate nucleotides. Nucleotide excision repair has been extensively studied in bacteria.
In E. coli, recognition and excision of the damage is carried out by a group of proteins encoded by the uvrABC and uvrD genes. The protein products of the uvrA, uvrB and uvrC genes function together as the so-called UvrABC excinuclease. The damage is initially recognized and bound by a complex of the UvrA and UvrB proteins. Once the complex is bound, the UvrA dissociates, leaving the UvrB attached to the DNA, where it is then joined by the UvrC protein.
Strand nicking
It is the complex consisting of UvrB and C that acts to cut the phosphodiester backbone on either side of the damage, creating nicks in the strand about 12-13 nucleotides apart. A helicase encoded by uvrD then unwinds the region containing the damage, displacing it from the double helix together with UvrBC. The gap in the DNA is filled in by DNA polymerase, which copies the undamaged strand, and the nick is sealed with the help of DNA ligase.
Nucleotide excision repair is also an important pathway in eukaryotes. It is particularly important in the removal of UV damage in humans, given that we lack photolyases. A number of proteins have been identified that function in ways similar to the Uvr proteins.
The importance of these proteins is evident from the fact that mutations in the genes that encode them can lead to a number of genetic diseases, like Xeroderma pigmentosum, or XP. People with XP are extremely sensitive to UV exposure, because the damage caused by it cannot be repaired, leaving them at a much higher risk of developing skin cancer.
Two repair modes
Nucleotide excision repair operates in two modes, one known as global genomic repair and the other as transcription-coupled repair. While the function of both is to remove helix-destabilizing damage like cyclobutane pyrimidine dimers or chemical adducts, the way in which the lesions are detected differs.
In global genomic repair, damage is identified by surveillance of the entire genome for helix distorting lesions. In the case of transcription-coupled repair, the stalling of the RNA polymerase at a site of DNA damage is the indicator that activates this mode of nucleotide excision repair.
Base excision repair
Base excision repair (BER) is a repair mechanism that deals with situations like the deamination of cytosine to uracil (Figure 7.43) or the methylation of a purine base. These changes do not typically distort the structure of the DNA helix, unlike chemical adducts or UV damage.
In base excision repair a single damaged base is first removed from the DNA, followed by removal of a region of the DNA surrounding the missing base. The gap is then repaired.
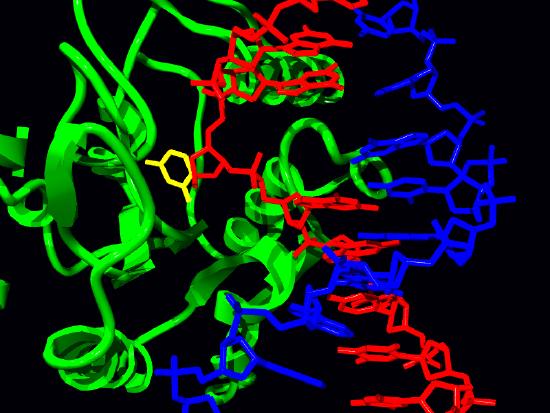
Uracil-DNA glycosylase
The removal of uracil from DNA is accomplished by the enzyme uracil-DNA glycosylase that can recognize uracil in DNA and break the glycosidic bond between the uracil and the sugar in the nucleotide (Figure 7.44). The removal of the base leaves a gap called an apyrimidinic site (AP site) because, in this case, uracil, a pyrimidine was removed. It is important to remember that at this point the backbone of the DNA is still intact, and the removal of a single base simply creates a gap like a tooth that has been knocked out.
The formation of the AP site triggers the activity of an enzyme known as an AP endonuclease that cuts the DNA backbone 5’ to the AP site. In the remaining steps, a DNA polymerase binds to the nick, then using its exonuclease and polymerase activities, replaces the sequence in this region. Depending on the situation, a single nucleotide may be replaced (short patch BER) or a stretch of several nucleotides may be removed and replaced (long patch BER). Finally, as always, DNA ligase acts to seal the nick in the DNA.
Repair of double-strand breaks
While all the repair mechanisms discussed so far fixed damage on one strand of DNA using the other, undamaged strand as a template, these mechanisms cannot repair damage to both strands. What happens if both strands are damaged? Ionizing radiation, exposure to certain chemicals, or reactive oxygen species generated in the cell can lead to double-strand breaks (DSBs) in DNA.
DSBs are a potentially lethal form of damage that, in addition to blocking replication and transcription, can also lead to chromosomal translocations, where part of one chromosome gets attached to a piece of another chromosome. Two different cellular mechanisms exist that help repair DSBs (Figure 7.45), homologous recombination (HR) and non-homologous end joining (NHEJ).
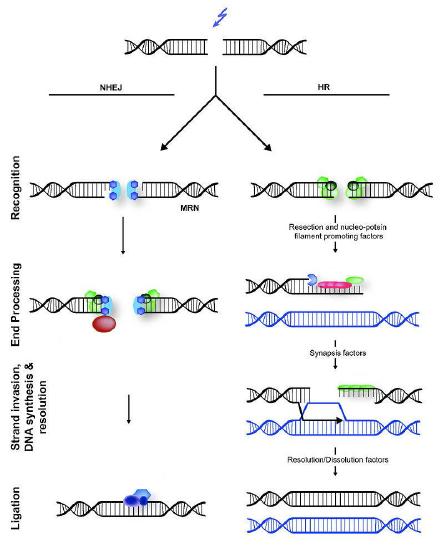
Figure 7.45 - Non-homologous end joining (left) versus homologous recombination (right) - Wikipedia
Homologous recombination repair commonly occurs in the late S and G2 phases of the cell, when each chromosome has been replicated and information from a sister chromatid can be used as a template to achieve error-free repair. Note that in contrast to excision repair, where the damaged strand was removed and the undamaged sister strand served as the template for filling in the damaged region, HR must use the information from another DNA molecule, because both strands of the DNA are damaged in DSBs.
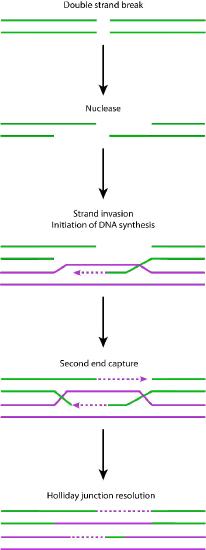
Nuclease action
Detection of the double-strand break triggers nuclease activity that chews back one strand on each end of the break. This results in the production of single-stranded 3’ overhangs on each end. These single-stranded ends are bound by several proteins, creating a nucleoprotein filament that can then “search” for homologous (matching) sequences on a sister chromatid.
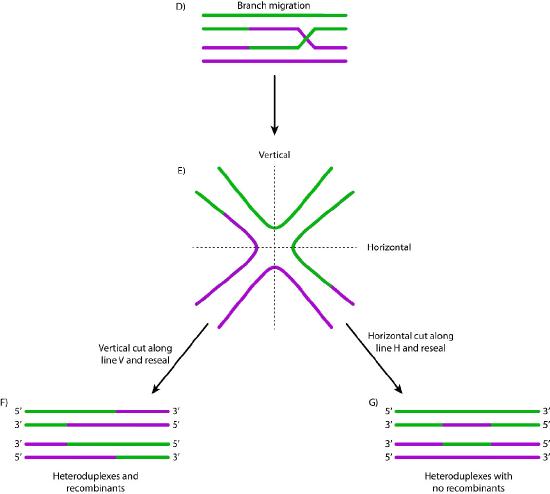
When such sequences are found, the nucleoprotein filament invades the undamaged sister chromatid, forming a crossover. This creates heteroduplexes made up of DNA strands from different chromatids. Strand invasion (Figure 7.47) is followed by branch migration, during which the Holliday junction moves along the DNA, extending the heteroduplex away from the original site of the crossover (Figure 7.48). In E. coli, branch migration depends on the activity of two proteins, RuvA and RuvB. The resulting recombination intermediate can be resolved, with the help of RuvC to give complete, error-free strands.
Non-homologous end joining
In contrast to homologous recombination, Non-homologous end joining (NHEJ) is error-prone. It does not use or require a homologous template to copy, and works by simply chewing back the broken ends of DSBs and ligating them together. Not surprisingly, NHEJ introduces deletions in the DNA as a result.
Translesion DNA synthesis
As we have seen, cells have a variety of mechanisms to help safeguard the integrity of the information in DNA. One measure of last resort is translesion DNA synthesis, also known as bypass synthesis. Translesion synthesis occurs when a DNA polymerase encounters DNA damage on the template strand, but instead of stalling or skipping past the damage, replication switches to an error-prone mode, ignoring the template and incorporating random nucleotides into the new strand. In E.coli, translesion synthesis is dependent on the activities of proteins encoded by the umuC and umuD genes. Under the appropriate conditions (see SOS response, below) UmuC and UmuD are activated to begin bypass synthesis. Being error-prone, translesion synthesis gives rise to many mutations.
The SOS response
Named for standard SOS distress signals, the term “SOS repair” refers to a cellular response to UV damage. When bacterial cells suffer extensive damage to their DNA as a result of UV exposure, they turn on the coordinated expression of a large number of genes that are necessary for DNA repair. These include the uvr genes needed for nucleotide excision repair and recA, which is involved in homologous recombination. In addition to these mechanisms, which can carry out error-free repair, the SOS response can also induce the expression of translesion polymerases encoded by the dinA, dinB and umuCD genes.
How are all these genes induced in a coordinated way following UV damage? All of the genes induced in the SOS response are regulated by two components. The first is the presence of a short DNA sequence upstream of their coding region, called the SOS box. The second is a protein, the LexA repressor (Figure 7.49), that binds to the SOS box and prevents transcription of the downstream genes. Expression of the genes requires the removal of LexA from its binding site. How is this achieved?
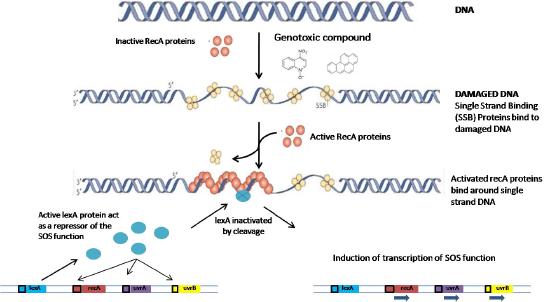
When exposure to radiation results in DNA breaks, the presence of single-stranded regions triggers the activation and binding of RecA proteins to the single-stranded region, creating a nucleoprotein filament. The interaction of the RecA with the LexA repressor leads to autocleavage of the repressor, allowing the downstream gene(s) to be expressed (Figure 7.50).
The genes controlled by the LexA repressor, as mentioned earlier, encode proteins that are necessary for accurate DNA repair as well as error-prone translesion synthesis. The various genes involved in DNA repair are induced in a specific order. In the initial stages, the repair genes that are derepressed are for nucleotide excision repair, followed by homologous recombination, both error-free mechanisms for repair. If the damage is too extensive to be repaired by these systems, error-prone repair mechanisms may be brought into play as a last resort.
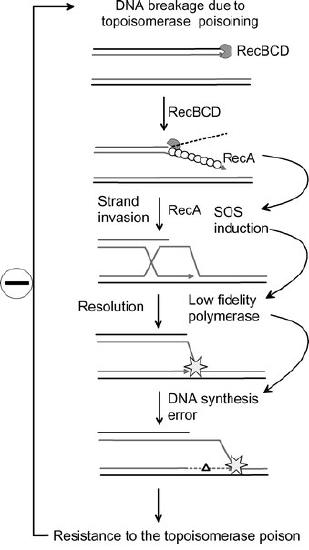
SOS response and antibiotic resistance
The increased mutation rate in the SOS response may play a role in the acquisition of antibiotic resistance in bacteria (Figure 7.51).
An example is the development of resistance to topoisomerase poisons like the fluoroquinolone family of drugs. Fluoroquinolones inhibit the ability of topoisomerases to religate the ends of their substrates after nicking them to allow overwound DNA to relax. This results in accumulation of strand breaks that can trigger the SOS response. As a consequence of error-prone DNA synthesis by low fidelity polymerases during the SOS response, there is a large increase in the number of mutations. While some mutations may be lethal to the bacteria, others can contribute to the rapid development of drug resistance in the population.