
1.6: Preparing DNA Template for Mutant RNA Sensor Synthesis Using a Restriction Endonuclease
- Page ID
- 18923

6.1 Learning Objective
In this lab, you get our DNA template containing the mutant sensor RNA ready for RNA synthesis. With this process, you will learn how restriction endonucleases – enzymes that cleave DNA at a given sequence – achieve their extraordinary specificity.
6.2 Mini Project Flowchart
The bolded block in the flowchart below highlights the role of the current experiment in the mini project.

6.3 Synopsis
The goal of the mini project was to identify nucleotides in the toxin sensor that are essential for recognizing the antibiotic tetracycline. In the quest toward this goal, you identified evolutionarily conserved elements in the sensor and modified them. You designed primers to change the sequence of the sensor and made mutant sensor DNAs using PCR amplification and plasmid preparation. To test whether these mutant sensors are capable of recognizing tetracycline you have to synthesize the mutant sensor RNAs. Recall, the sensor is an RNA molecule, but its sequence is coded in the genome at the DNA level. RNA is synthesized using a linear DNA template and an RNA polymerase. Therefore, the plasmid DNA that codes for the mutant sensor has to be cut (linearized) using a restriction endonuclease. The plasmid vector is cleaved at the end of the toxin sensor sequence to prevent the RNA polymerase from transcribing the entire plasmid vector into RNA (you only need the sensor). RNA polymerases work similarly to DNA polymerases and you will learn more about them in detail during the next lab.
6.4 How do Restriction Endonucleases Work?
Restriction endonuclease evolved to cleave intruder DNA, but leave the DNA of the host organism intact. They are used extensively by molecular biologists as a tool to specifically cleave plasmid or in vitro synthesized DNA during cloning. Restriction endonucleases are enzymes that cut dsDNA at a specific sequence with million-fold specificity. This means that restriction endonucleases favor their target sequence million-fold over other sequences. Cognate DNAs have the specific sequence and are cleaved efficiently whereas non-cognate DNAs do not have the specific sequence, and therefore are not cleaved efficiently by the restriction enzyme. Restriction enzymes use a metal-activated water molecule as nucleophile. The metal-bound water molecule is more acidic than a regular water molecule. Therefore, the metal-bound water loses its proton more easily and becomes a hydroxyl group. A hydroxyl group is a strong nucleophile and able to catalyze cleavage of the phosphodiester bond very efficiently (Fig. 6.1).
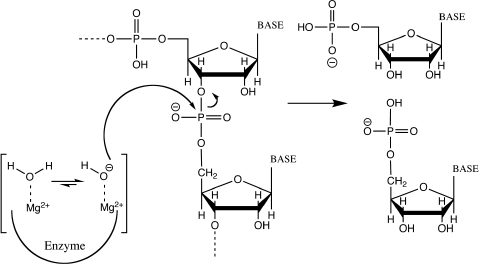
6.5 How do Restriction Enzymes Achieve Million-Fold Specificity?
Restriction endonucleases favor their cognate sequence million fold over other sequences. How is this extraordinary specificity achieved? Restriction endonuclease cleavage sites have twofold symmetry. Likewise, restriction enzymes are dimers and have complementary twofold symmetry (Fig. 6.2).

The restriction endonuclease forms a more extensive H-bonding network with its cognate than with its non-cognate DNA. As a result, the cognate DNA is bent (it has a kink in the dsDNA structure) and the scissile bond (the bond that is cleaved) is placed right in the middle of the enzyme’s active site for cleavage. The energy required to bend the cognate DNA comes from the extensive H-bonding between restriction enzyme and the cognate DNA (Fig. 6.3; Fig 6.4).


6.6 How Do We Judge Whether The Plasmid DNA is Successfully Linearized?
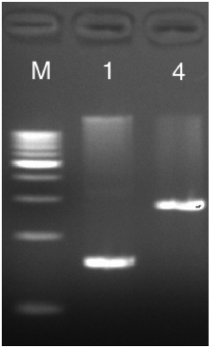
Agarose gel electrophoresis is a powerful technique to separate DNA based on size and/or shape (see Chapter 5.). Since plasmid DNA coding for the mutant sensor RNA was synthesized by bacteria, it is supercoiled and compact. Once this DNA is cut with a restriction endonuclease, it is nicked and hence has a more elongated shape. As a result supercoiled (uncut) plasmid DNA migrates farther in the agarose gel than linearized (cut) DNA. If the DNA linearization is successful, the sample containing the cut DNA should have a single band that migrates less far in the gel than the uncut DNA sample (Fig. 6.5).
6.7 What are We Going to do in the Lab?
First, you will digest the plasmid DNA coding for the mutant toxin sensor with the restriction enzyme BamHI and evaluate the success of the reaction using agarose gel electrophoresis. While the gel is running, you will remove the restriction enzyme by phenol-chloroform extraction and increase DNA concentration by ethanol precipitation.
PROTOCOLS
Reagents and equipment needs are calculated per six student teams. There is ~20% excess included.
Equipment/glassware needed
- Three 100 ml corked Erlenmeyer flask
- Three 100 ml graduated cylinder
- Three sets of micropipettes (20-100 μl and 2-20 μl)
- Gel documentation system
- Water bath (for restriction digest) set to 37 °C
- Thirty 1.5 ml centrifuge tubes (Eppendorf)
- 1 microcentrifuge capable of 10,000g
Solutions needed
- 3 l 1 x TAE (Tris-acetate-EDTA) pH=8.0
- 30 ml 1% EtBr or equivalent nucleic acid stain
- 20 ml agarose gel loading dye (6x, Biorad)
- 30 ml 1 kb molecular weight ladder (Biorad)
- 2 ml Phenol:Chlorophorm:isoamyl alcohol mix
- 150 ml 3M Na-acetate, pH=5.2
- 1.5 ml isopropanol
Restriction digestion of pUC19-ykkCD plasmid vector
Each member of the team performs a digestion
Mix the following solutions in a centrifuge tube
- 5 μl buffer 3 (New England Biolabs)
- 5 μl 1 mg/ml bovine serum albumin solution
- 5 μl BamHI restriction enzyme (New England Biolabs)
- 75 μl plasmid DNA
Incubate in 37 °C water bath for 1 hr.
Agarose gel electrophoresis
- Weight 1 g agarose and place it to the corked Erlenmeyer flask provided.
- Add 100 ml 1xTAE buffer using a graduated cylinder.
- Heat for 1 min in microwave, swirl flask and briefly cool under water.
- Add 10 μl EtBr. (Wear gloves. EtBr is mutagenic.) Swirl flask for mixing.
- Pour liquid into gel cassette and position combs. It takes about 20 min for an agarose gel to set.
- Once the gel is set load your samples with a molecular weight ladder. Gel should be run for 20 min with 100 V.
- Take picture of the gel using a gel documentation system.
Load the 5 ml of the following samples mixed with 1ml loading dye onto the agarose gel
- Molecular weight marker
- Plasmid DNA from the plasmid prep (uncut DNA sample)
- Plasmid DNA digested with the restriction endonucleases (cut DNA)
Phenol-chloroform extraction and ethanol precipitation
- Add 100 μl phenol/chloroform mixture to your sample.
- Vortex for 20 sec.
- Spin for 2 min in a microcentrifuge at maximum speed.
- Discard bottom layer. (This step removes the organic layer containing the restriction endonuclease.)
- Add 100 μl chloroform, vortex and spin as before. Remove bottom layer. This step removes residual phenol.
- Add 10 μl 3 M NaAc and 100 μl isopropanol to your mixture.
- Vortex sample and place into -20 ºC freezer for precipitation.
Notes to the instructor
The experiment in Chapter 6 is designed to teach students how to perform enzymatic cleavage of the pUC19-ykkCD plasmid vector using BamHI restriction enzyme. The same protocol with minimal modifications may be used with a different DNA and/ or restriction endonuclease. BamHI restriction enzyme was purchased from New England Biolabs, but other vendors may be used. Two student teams shared one agarose gel to limit equipment need while maximizing exposure to this important technique. If the number of equipment is limited six student teams can easily share one agarose gel apparatus. Since restriction endonuclease digest takes an hour it is convenient to start the laboratory session by setting up the reactions and giving pre laboratory lecture while the reactions are running. This laboratory has two breaks while the agarose gel sets and runs. These times can be utilized to complete either the restriction endonuclease or electrophoresis worksheet.
Prelab Questions for DNA Linearization
Define the following terms.
- Restriction endonuclease

- Scissile bond
- Cognate DNA and noncognate DNA

- To test DNA quality you need to resolve the DNA sample on an agarose gel. You need 1 l 1 x TAE buffer (Tris-acetate EDTA) to make and run an agarose gel. You purchased 40 x concentrated TAE buffer stock. Describe how you make the 1 l 1 x TAE solution needed for the gel (show your calculation).

DNA Linearization
Lab Report Outline and Point Distribution
1. Several sentences defining the goal/purpose of this experiment. Describe the significance of this experiment within the mini project. (3 pts.)
2. Briefly define/describe a restriction endonuclease. Explain how specificity is achieved. (4 pts.)
3. Include a copy of your agarose gel scan with your samples marked. (4 pts.)
4. Report the concentration of your plasmid DNA. (2 pts.)
5. Evaluate plasmid DNA yield based on the DNA concentration (how many mg of DNA did you get). Show your yield calculation and comment on how good was your DNA yield. (6 pts.)
6. Evaluate plasmid DNA purity based on the agarose gel scan and the 260/280 reading determined by UV absorbance. Explain. (6 pts.)
7. Judge the success of your DNA linearization based on the agarose gel scan. Explain. (4 pts.)
8. Judge the size of your plasmid DNA provided that the DNA step ladder has sizes: 1000 bp, 2000 bp, 3000 bp, 4000 bp, 5000 bp, 6000 bp, 7000 bp, 8000 bp, 9000 bp, 10,000 bp. Explain. (4 pts.)
9. Electrophoresis Problem Set (17 pts.)
Restriction endonucleases are very specific and cleave DNA at specific nucleotide sequences. When these enzymes act on large DNA, it is broken down into a set of many, many fragments of different sizes. The size distribution of such fragments is essentially unique for each different DNA. This distribution provides a DNA “fingerprint” that is unique for each individual. DNA fingerprints have been used to identify genetic mutations, to trace hereditable traits, to establish parentage, and to place suspects at the scene of a crime.
There are several different techniques that give rise to a DNA fingerprint. Some of the problems below will describe techniques and ask you to interpret the results.
1. (3 pts.) There are thousands of different restriction endonucleases, each with its own specificity. For example, BamHI, EcoRI, and XhoI have the following specificities (the cleavage sites are marked with arrows):

Identify the cleavage sites on the following DNA (mark each site with a line and the name of the specific endonuclease):
5'CCCGAGGATCCTTAGGAATTCATCTA3'
3'GGGCTCCTAGGAATCCTTAAGTAGAT5'
2. (4 pts.) Show the sequence of the product fragments from the BamHI cleavage. Why are these products said to have “sticky ends?”
3. (6 pts.) Restriction fragment length polymorphism (RFLP, often pronounced “riflip”) is the oldest form of DNA fingerprinting. It depends on the fact that the DNA from each individual has many segments with unique sequences. Each segment can have a variety of sequences that are inheritable. This characteristic is termed “polymorphism.” Polymorphism means that each individual have DNA with different restriction enzyme cleavage sites. Briefly, RFLP involves
- Treating the DNA with a specific restriction endonuclease;
- Separating the resulting fragments by size on an agarose gel;
- Visualizing specific fragments using complementary DNA probes.
The following agarose gel was used to establish parentage. It was generated using single locus RFLP. That is the probe used was sensitive to only one polymorphic segment in the DNA.
To interpret these results you need to know
– Each individual carries two copies of DNA and, meaning, there are two different polymorphisms for each DNA segment;
– Each individual inherits one DNA copy from each parent.

http://www.biology.arizona.edu/)
a. Are there any children that don’t appear to be biological children of these parents? Explain.
b. What must be true of the child’s DNA fingerprint in order to establish that he/she is related to the mother and father?
4. (4 pts.) In the following DNA fingerprints, C/AF Mix is a lane that contains both the alleged father’s DNA as well as the child’s DNA.
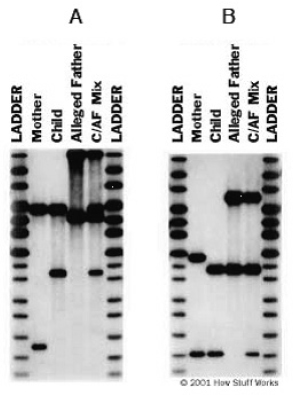
(Courtesy of Genelex Corporation)
Is paternity proven in either case? Briefly explain.
5. (6 pts.) PCR has been combined with RFLP to increase the sensitivity of the DNA fingerprinting. In this technique PCR amplifies a specific DNA segment of interest. Then, changes in the sequence are identified by RFLP.
For example, sickle cell anemia can be diagnosed using this technique. Sickle cell anemia is caused by a single mutation on the DNA that code for one of hemoglobin protein chains. This mutation also eliminates a restriction endonuclease cleavage site for the enzyme; Cvn. Loss of the cleavage site is then diagnostic for the sickle cell gene.
In the following, a mother, father and children have been tested for the sickle cell gene via DNA fingerprinting. The diagram above the electrophoresis pattern shows parents (row I) and children (row II). Females are symbolized by a circle and males by a square. The normal gene is indicated with an open symbol and the sickle cell gene by the filled symbol. (Remember each individual carries two copies of the gene.)
Each individual’s symbol corresponds to a lane in the electrophoresis gel. The lanes are also marked with “S” for presence of the sickle cell mutation and “A” for presence of the normal gene. (The DNA ladder is on the right and is given in number of bases. Note that the gel is “upside down” - the smaller fragments are closer to the top.)
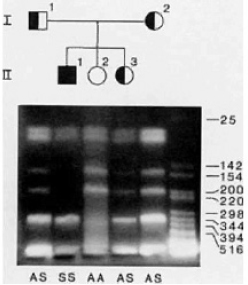
(Courtesy of Dr. Carole Ober)
- Estimate the size of the cleavage fragments that are only present in the individual without the sickle cell mutation. Briefly explain your choice.
- From what band do these cleavage fragments come? Can you identify the band and estimate its size? Explain your reasoning.
- The researcher diagnosed these subjects (SS, AS, or AA) based on this electrophoresis. Do you see any inconsistencies in the diagnosis? Briefly explain.
Design primers for conventional (cutting and pasting) mutagenesis to place the ykkCD toxin sensor into the cloning vector pUC19. Experiments like this are usually the first step in studying a protein or RNA in the lab. The sequence of the toxin sensor and the map of the cloning vector are given below.
DNA sequence of ykkCD sensor:
Tgtaaagttttctagggttccgcatgtcaattgacatggactggtccgagagaaaacacatacgcgtaaataga
agcgcgtatgcacacggagggaaaaaagcccgggagag
Map of the pUC19 cloning vector

The ykkCD toxin sensor has to be inserted into pUC19 using a part of the cloning vector called multiple cloning site or polylinker MCS. This part of the cloning vector contains cut sites for several restriction enzymes and designed to allow insertion of DNA sequence. In case of pUC19 the available restriction enzymes are: EcoRI, SacI, KpnI, SmaI, BamHI etc.
To complete the task of pasting the ykkCD sequence into pUC19 answer the questions below:
1. Step one in a conventional cloning project is to choose a pair of restriction enzymes from the ones in the polylinker MCS that do not cleave the sequence of interest (the ykkCD sensor). Restriction enzymes that cut the ykkCD sequence cannot be used for cloning, because they would cleave the sequence of interest and would not allow pasting the entire sequence into the cloning vector. To select appropriate restriction endonucleases follow the two steps below.
a. Paste the sequence of the ykkCD sensor into a program called Webcutter (http://www.firstmarket.com/cutter/cut2.html). List the names of restriction enzymes that do not cut the ykkCD sequence.
b. Which enzymes in the MCS polylinker may be used to perform your cloning project? Provide a brief explanation.
2. Design primers to perform the cloning experiment.
a. Provide the top and bottom primer sequences. Briefly explain how you designed these primers. Which part of the DNA these primers anneal to? Remember, there is a difference between Quickchange and conventional PCR.
b. Calculate the TM (melting temperature) for each primer. You may use Oligo calculator (http://www.basic.northwestern.edu/biotools/oligocalc.html) to predict TM.
c. Conventional PCR requires addition of the special sequence that is recognized by each restriction enzyme called restriction site. You may find the recognition sequence for each restriction enzyme at http://www.neb.com.
Your revised primer sequences should follow this pattern
Restriction site sequence + primer sequence from “b”.
d. Write the sequence of both primers in the 5’ to 3’ direction.