
1.5: Purifying Mutant Toxin Sensor DNA from Bacterial Cells and Evaluating its Quality Using Agarose Gel Electrophoresis and UV Spectroscopy
- Page ID
- 18922

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)5.1 Learning Objective
Last week you generated the plasmid DNA containing the mutant toxin sensor using QuickChange PCR amplification. This week you will learn how to extract and purify this plasmid DNA from E coli cells, and how to check the quality of the plasmid DNA using agarose gel electrophoresis and UV spectroscopy.
5.2 Mini Project Flowchart
The bolded block in the flowchart below highlights the role of the current experiment in the mini project.

5.3 Purification of Plasmid DNA from Bacterial Cell (Plasmid Prep)
Last week, you used PCR amplification to synthesize plasmid DNA containing the mutant toxin sensor. PCR amplification is an excellent tool to change the sequence of the DNA, but it does not generate sufficient amount of plasmid DNA. PCR amplification typically yields a few hundreds of nanograms of DNA. While cloning, transfection or RNA synthesis generally requires DNA at the microgram or even milligram level. Also, plasmid DNA generated by PCR amplification is circular while plasmid DNA synthesized by bacteria is mostly supercoiled. Supercoiled DNA has the same molecular weight as its circular counterpart, but it is more compact. DNA in the supercoiled conformation is smaller, and it is better protected against harm caused by restriction endonucleases or environmental hazards (Fig 5.1).

For these reasons, PCR amplification is usually not used to make large quantities of DNA. To synthesize DNA for most applications in chemistry and biology, bacterial cells have to be used. During the plasmid prep, we use the DNA synthesis regime of bacterial cells, typically E. coli, to prepare the plasmid DNA for us. In other words, we hijack E. coli to synthesize our DNA.
The steps of plasmid vector synthesis using bacterial cells are as follows:
- Transform plasmid DNA into the bacterial cell.
- Grow bacteria containing the desired plasmid (5 to 500 ml).
- Purify plasmid DNA from bacterial cells.
5.4 Transformation
In order to use bacterial cells to synthesize plasmid DNA, the plasmid DNA has to be placed inside the bacterial cell; meaning we have to force bacteria to take up the plasmid DNA. This is problematic, because the bacterial cell membrane is apolar and represents an impermeable barrier to polar molecules like DNA. Transformation pokes “holes” on the cell membrane to allow DNA entry. Two methods are available for transformation: heat shock and electroporation. Heat shock involves quick heating and cooling of bacterial cells that were previously treated with CaCl2. As cell membranes are permeable to Cl- ions, once Cl- goes through as a hydrated anion, it leads to “swelling of the cell”. The 42 °C heat shock leads to formation of holes (pores) on the membrane that allows DNA entry. During electroporation, these holes are generated via a brief electroshock.
Transformation is a low-yield procedure: relatively few cells take up the plasmid, despite the heat shock or electroporation treatment.
5.5 Cell Growth
As a result of the low yield, a selective marker has to be used to distinguish cells that harbor the plasmid from cells that do not. The most commonly used selective marker with bacterial systems is the usage of an antibiotic. Plasmids harbor a resistance gene for a given antibiotic. This means cells that took up the plasmid will grow on media containing the antibiotic, while cells that did not take up the plasmid will not. Consequently, plating the transformation mixture on solid medium that contains the selective antibiotic, ampicillin in our case, will only yield colonies from bacteria that harbor the plasmid containing the ykkCD sensor RNA.
Once E coli cells that harbor the plasmid DNA containing the mutant ykkCD sensor are identified, these cells are inoculated into liquid medium containing the selective antibiotic (ampicillin) and grown for 16 hrs. at 37 °C with vigorous shaking.
5.6 Purification of Plasmid DNA from Bacterial Cells
When plasmid DNA is synthesized via PCR amplification, the resulting DNA is very clean. However, when DNA is synthesized using bacterial cells the plasmid DNA is contaminated with bacterial cell components: their cell membrane, cellular proteins, DNA, and RNA. During the plasmid prep, all of these contaminants have to be removed. Plasmid preps utilize the fact that DNA is highly negatively charged (compared to proteins), very soluble in water (compared to lipids and membrane proteins) and fairly small (compared to genomic DNA).
Make sure you follow the plasmid prep protocol exactly as written. The first few steps of plasmid purification (from cell breakage to loading the DNA onto the purification column) significantly impact the purity and quantity of the plasmid DNA.
The exact protocol of plasmid purification is dependent of the kit used and will be provided in lab. A general outline of procedure is presented below.
- Plasmid preps began with breaking the bacterial cell membrane (lysis) to free the plasmid DNA. This step is usually achieved by an alkaline lysis. Cell breakage can also be achieved using the enzyme lysozyme. Be careful to NOT allow cell lysis to proceed longer than instructed. Handle cell lysate gently to prevent shearing of chromosomal DNA. This could cause contamination of the plasmid DNA with genomic DNA.
- After cell lysis, the plasmid DNA will be in solution with soluble proteins, but anything that is not water-soluble, the genomic DNA, lipids and membrane proteins, will form sediment. This sediment can either be removed by centrifugation or filtration. Either way, this step purifies the plasmid DNA from proteins, lipids, and genomic DNA.
- The plasmid DNA is further purified via column purification. Often a silica-based anion-exchange chromatography column is used. This step takes advantage of the negative charge of DNA to separate the plasmid DNA from the host’s genomic DNA. Since genomic DNA is much larger it has a higher negative charge then plasmid DNA, it interacts more strongly with the anion exchange column. As a result genomic DNA remains bound to the anion exchange column while the plasmid DNA is eluted.
5.7 Agarose Gel Electrophoresis
To evaluate the quality of the plasmid DNA agarose gel electrophoresis will be used. Electrophoresis is a technique that is used to separate molecules based on charge and shape. Separation is achieved by moving the negatively charged molecules like DNA through an electric field from the negative to the positive electrode. Shorter molecules migrate faster than longer ones. To prevent spreading of the sample during electrophoresis a solid medium is used like agarose or polyacrylamide. Agarose is a polysaccharide that is generally extracted from seaweed and is used to separate large nucleic acids (> 500 base pair). The pore size of the polymer is dependent on the percentage of the agarose used (0.6 – 2%). Higher percentage leads to smaller pore size and is designed to separate smaller molecules. Agarose gels generally do not denature (unfold) DNA. Therefore, DNA conformation also affects the rate of migration.
The rate of migration in an agarose gel is described by the equation below where v is velocity (rate), q is the charge of the molecule, E is the strength of the electric field and f is friction.

DNA migrating through a gel with smaller pore size experiences more friction and travels less distance than the same molecule migrating through a gel with larger pore size. Likewise, DNA with more elongated shape travels smaller distance in the gel than compact DNA, because compact DNA experiences less friction as it migrates through the gel. Therefore, supercoiled DNA (more compact) migrates faster in an agarose gel than nicked or circular DNA (more elongated shape). DNA with higher molecular weight travels less far than DNA with lower molecular weight, as long as their shape is similar. The relationship between shape, molecular weight and distance traveled in an agarose gel is seen in Fig. 5.2.
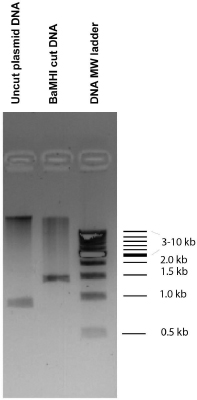
Nucleic acids are visualized on an agarose gel using ethidium bromide (EtBr) staining. EtBr is fluorescent and it interacts with the nucleobases in DNA (intercalates with DNA bases). Excess of the EtBr migrates out of the gel quickly due to its small size. When exposed to UV light nucleic acids “light up” in an agarose gel due to EtBr fluorescence. SYBR green nucleic acid stains work in a similar fashion (Fig 5.3).

5.8 Application of Agarose Gel Electrophoresis
Agarose gel electrophoresis is a powerful technique that is used to separate nucleic acids by size and/or shape. It has both preparative and analytical applications. As an analytical tool, agarose gel electrophoresis can be used to evaluate the success of any enzymatic reaction that results in significant change of DNA shape, such as cleavage by restriction endonucleases (see more detail in Chapter 6). Restriction endonucleases are enzymes that cleave DNA, and thus turn supercoiled DNA (fast migrating) into linear DNA (slow migrating). As a result, the success of a cleavage reaction by restriction endonuclease can be judged by how far the cleaved DNA sample migrates on an agarose gel as compared to the original (uncut) DNA. Fig 5.2 compare lanes with cut and uncut DNAs. A preparative application of agarose gel electrophoresis is to remove any uncut (supercoiled) DNA from a sample treated with a restriction endonuclease. In this case, the slow migrating band (cut DNA) is excised from the agarose gel and the DNA is eluted from the gel piece using commercially available kits.
5.9 DNA Quality Control Using UV Spectroscopy
UV spectroscopy is commonly used to determine the concentration of plasmid DNA. Nucleic acids strongly absorb UV light at 260 nm, due to the nucleobases (heterocyclic aromatic compounds). The concentration of DNA produced is estimated using the formula below:
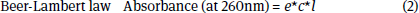
Where e is the absorption coefficient (20 ml/mg*cm for plasmid DNA), l is the path length (1 cm) and c is the concentration of the plasmid DNA in mg/ml. By measuring the absorbance of the plasmid DNA produced, the concentration can be calculated.
PROTOCOLS
Reagents and equipment needs are calculated per six student teams. There is ~20% excess included.
Equipment/glassware needed
- Three 100 ml corked Erlenmeyer flask
- Three 100 ml graduated cylinder
- Three sets of micropipettes 20-100 µl and 2-20 µl.
- UV spectrometer
- Gel documentation system
- Water bath (for transformation)
- Orbital shaker (for cell growth)
- Six 10-15 ml tubes (sterile)
- Six 1.5 ml centrifuge tubes (Eppendorf)
Solutions needed
- 3 l 1 x TAE (Tris-acetate-EDTA) pH=8.0
- 30 ml 1% EtBr (or equivalent nucleic acid stain)
- 20 ml Agarose gel loading dye (6x, Biorad)
- 30 ml 1 kb molecular weight ladder (Biorad)
- 30 ml LB (Luria-Bertani) broth and six LB/Ampicillin (100 mg/ml) plates
- 30 ml 100 mg/ml (1000 x) Ampicillin (for cell growth)
Transformation (may be done by teaching assistants ahead of time)
- Set water bath to 42 °C.
- Label 1 sterile glass or plastic tube and 1 microcentrifuge tube for each mutant.
- Label 1 LB-agar plate containing 100 mg/ml ampicillin for each mutant.
- Thaw E. coli Dh5a competent cells on ice (100 ml competent cell is used for each mutant).
- Add 10 μl PCR amplified mutant DNA to each tube containing competent cells. Keep cells on ice for 20 min.
- Heat shock DNA as follows:
- Place cell into 42 °C for 2 min.
- Move cells to ice for 3 min.
- While performing the heat shock treatment pipette 900 μl LB broth into previously labeled sterile tubes and place them into 37 °C orbital shaker for pre-warming.
- Add heat shocked cells to tubes containing 900 μl pre-warmed LB broth.
- Grow cells while shaking gently (120 rpm) for 1 hr.
- Transfer cells into previously labeled centrifuge tubes. Pellet cells for 2 min in a microcentrifuge using 5000 rpm.
- Remove supernatant. Resuspend cells in 100 μl LB broth and plate then on the previously labeled LB/ampicillin plates.
- Grow cells overnight in 37 °C incubator.
Cell growth (may be done by teaching assistants ahead of time)
- Label 1 sterile glass tube for each mutant.
- Inoculate a single colony into 1 ml LB/ broth supplemented with 100 μg/ml ampicillin. Grow cells for 5-6 hrs. in an orbital shaker at 37 °C with vigorous shaking (280 rpm).
- Label 1 sterile 120 ml Erlenmeyer flask for each mutant.
- Use 100 μl cells from “step 2” to inoculate 40 ml LB broth supplemented with 100 μg/ml ampicillin for each mutant.
- Grow these cells overnight with vigorous shaking (280 rpm).
- Cells may be harvested (10 min centrifugation with 5000 x g; remove supernatant) and stored at -20 °C.
Plasmid purification from E. coli cells
Protocol depends on kit used and will be provided in lab. See pg 47 for a basic overview.
Agarose gel electrophoresis
- Weight 1 g agarose and place it to a corked Erlenmeyer flask provided.
- Add 100 ml 1 x TAE buffer using graduated cylinder.
- Heat for 1 min in microwave, swirl flask and briefly cool under water.
- Add 10 μl EtBr (Wear gloves. EtBr is mutagenic); swirl flask for mixing.
- Pour liquid into gel cassette and position combs. It takes about 20 min for the agarose gel to set.
- Once gel is set load your sample plus 10 μl molecular weight ladder (with 2 μl loading dye). Run gel for 20 min with 100 V.
- Take picture of gel using a gel documentation system.
UV spectroscopy (this protocol is for Nanodrop spectrometer; for conventional UV spectrometer large volumes of DNA need to be used)
- Turn on spectrometer and choose nucleic acid assay setup.
- Blank spectrometer with 2 μl Millipore water.
- Measure the absorbance of a water sample to ensure that the spectrometer is clean.
- Use 2 μl plasmid DNA sample to measure the concentration of your plasmid DNA.
- Record DNA concentration in mg/ml.
Notes to the instructor
The experiment in Chapter 5 is designed to isolate plasmid DNA containing mutant ykkCD tetracycline sensor RNA using E. coli cells. The same protocol with minimal modification could be used to perform plasmid purification of any DNA from E. coli cells. At the authors’ institution, students were provided with liquid cell cultures that contained the mutant toxin sensor DNA. Transformation and cell growth was performed by teaching assistants in the absence of students. This setup saves lab time, but somewhat limits students’ understanding of the steps required to make plasmid DNA. If desired, a separate lab session may be allocated where students perform transformation and cell growth. Any commercially available E. coli Dh5α competent cell is suitable for transformation. The E. coli strain and the protocol to make them competent for transformation used by the authors is available upon request. Plasmid Midi Kit from Qiagen works well to purify plasmid DNA, but similar kits from other vendors may work well. A mid-size plasmid purification kit is recommended to ensure that sufficient quantity of the plasmid DNA is produced.
Prelab Questions for Plasmid Prep
Define the following terms.
- Transformation

- Agarose gel

- Supercoiled DNA vs Linear DNA
- Beer-Lamber law. Define each term in the equation.
- What type of precautions do you have to observe while handling EtBr and why?

DNA Purification
Lab Report Outline and Point Distribution
1. Several sentences defining the goal/purpose of this experiment. Please indicate the role this step plays in the mini project (5 pts.).
2. Briefly describe the strategy used to purify plasmid DNA from cells (10 pts.). (What are the major contaminants? What properties of your plasmid DNA are exploited during purification and how? What are the differences between your plasmid DNA and other cellular macromolecules?)
3. Draw a detailed flowchart of the plasmid prep. Make sure you indicate the volume of each buffer that needs to be added and which fraction (soluble/insoluble or column/ filtrate) contains the plasmid DNA and which fraction is being discarded (20 pts.).
4. Define/describe transformation (5 pts.).
5. Define/describe the role the DpnI digestion plays in the Quickchange process (5 pts.).
Troubleshooting a plasmid prep
6. You colleague had significant genomic DNA contamination in their plasmid prep. In your opinion at which step(s) of the preparation a mistake was made? Briefly explain (5 pts.).
Electrophoresis Problem Set
1. (9 pts.) Describe the effects the following would have on a DNA agarose gel electrophoresis. Use the equation, v = qE/f, in a qualitative way as part of your explanation.
- The agarose concentration is increased from 1% to 1.5%.
- The voltage for the electrophoresis is decreased from 100 volts to 60 volts.
- The pH was changed from about neutral to acidic conditions (pH=3.0).
2. (5 pts.) You are asked to separate 1 μg of DNA on an agarose gel. The sample is to contain 1 x TAE buffer and the sample size is 10 μl. What volumes of the following would you use to prepare this sample: 0.5 μg/μl DNA stock solution, 10 x TAE buffer and water.
3. (9 pts.) An agarose gel was run to determine the size of several pieces of DNA. Lane “M” has a DNA size ladder of 11 steps (top 10 kb, 9 kb, 8 kb, 7 kb, 6 kb, 5 kb, 4 kb, 3 kb, 2 kb, 1 kb, 0.5 kb bottom). Measure the distances traveled to the nearest millimeter. Construct a plot of the log(kb) versus the distance traveled for the DNA ladder.
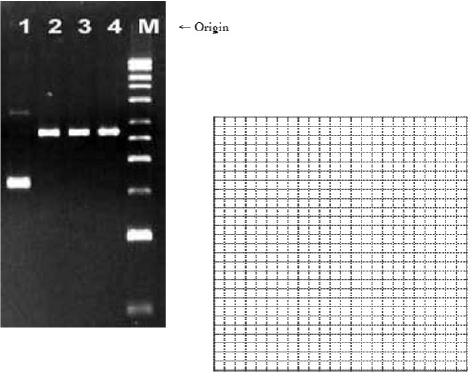
- (a) Using the plot, determine the size of the DNA (in kb) in lane 4 and lane 1.
- (b) The large band in Lane 1 is actually a circular plasmid DNA of the same size as the linear DNA in Lane 4. Briefly explain why the circular plasmid travels further in the gel than the linear DNA.