
1.4: Performing Site-Directed Mutagenesis
- Page ID
- 18921

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)4.1 Learning Objective
In this lab you will perform site-directed mutagenesis using the QuickChange mutagenesis kit (Stratagene). You will learn how polymerases work and how to amplify DNA using polymerase chain reaction (PCR).
4.2 Mini Project Flowchart
The bolded block in the flowchart below highlights the role of the current experiment in the mini project.

4.3 Review of Nucleic Acid Structure
Before you learn how polymerases work and the requirements of a successful PCR amplification you must review a few things about nucleic acid structure. Nucleic acids are polymers of nucleotides. The nucleotides are held together by phosphodiester bonds in the nucleic acid. Figure 4.1 shows the structure of a nucleotide. Nucleotides are made of a sugar moiety: ribose (RNA) or deoxyribose (DNA); a heterocyclic aromatic moiety: the nucleobase (A, C, G, T or U) and a phosphate group. When we refer to functional groups within the sugar moiety we use the apostrophe symbol; for example the second hydroxyl group on the ribose in the nucleotide is referred to as the 2'-OH group.
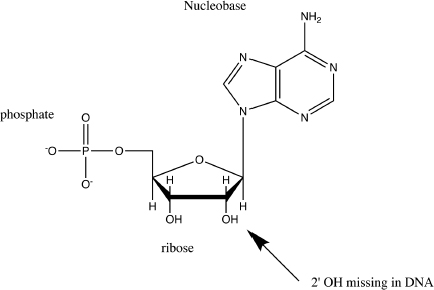
As we mentioned earlier, nucleotides are held together by the phosphodiester bond in the nucleic acid chain. The phosphodiester bond is made out of two phosphate ester bonds: each is formed between a OH group of the ribose or deoxyribose and a OH group of the phosphate. Since phosphoric acid is a moderately strong acid, the phosphodiester bond deprotonates under physiological conditions giving nucleic acids a negative charge (Fig. 4.2).

Nucleic acid chains have polarity just like protein chains. Nucleic acid polymers start with the 5’ phosphate of the first nucleotide and end with the 3'-OH group of the last nucleotide in the chain. Therefore the chain of the nucleic acid has a precise direction; it goes from 5' to 3' direction end just like protein chains go from the N-terminus to the C-terminus. In a double-stranded nucleic acid like DNA (our genetic material), one DNA polymer goes 5’ to 3’ and is called the top strand and the other goes from 3' to 5' direction is called the bottom strand. These two DNA strands are complementary to each other: adenine (A) against thymine (T) and cytosine (C) against guanine (G) to ensure proper Watson-Crick base pairing between the strands. In other words the two DNA strands in this double-stranded DNA (dsDNA) are complement of each other. Complement means that the two strands are complementary to each other and the chain direction is opposite: the top strand goes 5’ to 3’ whereas the bottom strand goes 3’ to 5’.
5’ AGGCCATTGGA 3’
3’ TCCGGTAACCT 5’
4.4 How do Polymerases Work?
Polymerases synthesize nucleic acids using a nucleic acid template. The sequence of the newly synthesized nucleic acid will be complement of the template sequence. During nucleic acid synthesis, the 3'-OH group of the growing nucleotide chain acts as a nucleophile to attack the phosphorous of the incoming nucleotide, a pyrophosphate (PPi) leaves and the phosphodiester bond forms. This means that the newly synthesized DNA chain grows in the 5’ to 3’ direction (Fig. 4.3).

To properly position the nucleophile for attack divalent metal ions (usually Mg2+) are necessary for successful DNA synthesis.
Polymerase mechanism in a nutshell:
– Polymerases synthesize DNA using a template that is the complement of the newly synthesized DNA.
– To synthesize DNA, polymerases require an OH group to act as a nucleophile. This OH comes from the growing nucleic acid chain. Recognize that the reaction is a nucleophilic substitution.
– The leaving group is pyrophosphate (PPi), which is a high-energy molecule that splits into two inorganic phosphates. This reaction is catalyzed by the enzyme inorganic phosphatase in vivo.
– Polymerization is energetically favorable, because two high energy anhydride bonds are broken (one in the incoming nucleotide triphosphate and the other in pyrophosphate) and one stable bond forms (ester bond connecting the nucleotides).
– To initiate DNA synthesis, DNA polymerases require a primer to provide the required OH group as nucleophile.
– Polymerases travel from 3' to 5' direction on the bottom strand of the dsDNA template while they synthesize the growing chain in the 5’ to 3’ direction.

4.5 Polymerase Chain Reaction (PCR) in Practice
To synthesize DNA in the lab we need to perform PCR amplification of the DNA of interest using a plasmid vector or genomic DNA as template. For successful PCR amplification we have to cycle through three steps 25-30 times. The steps of PCR amplification are as follows:
- Separation of the dsDNA template or strand separation
- Annealing of the primers to the template DNA (they form Watson-Crick base pairs) to initiate DNA synthesis
- DNA synthesis catalyzed by a polymerase
A graphic representation of a PCR cycle is seen on Fig. 4.5.
4.6 Why Did PCR Only Become Widely Available in the 1980s?
PCR amplification required two scientific breakthroughs. (1) Scientist had to invent a programmable machine that can cycle through the temperatures of the PCR cycle multiple times. The PCR machine can precisely and quickly change temperature; it can go from 95 ºC to 68 ºC within a few seconds. (2) Scientists had to find a DNA polymerase that can survive the first step of the PCR cycle: incubation at 95 ºC. This step is required to separate the strands of the dsDNA. Since polymerases are proteins, most bacterial or eukaryotic polymerases would not survive this incubation (think what happens when you boil an egg). Scientists turned to organisms that live under extreme conditions: in high temperature volcanic vents. Most of these organisms belong to the third kingdom of life – archaea. Polymerases from these organisms evolved to be active at 60-80 ºC, therefore incubation at 95 ºC for a few minutes does not harm them. Taq polymerase has a half-life of 2 hrs. at 95 °C while Pfu Turbo polymerase has a half-life of 19 hrs. at 95 °C. In addition, polymerase used for PCR has to be of high fidelity – this means that the polymerase very rarely incorporates a wrong nucleotide into the synthesized DNA (has low error rate). Taq polymerase has a 16% error rate in a 1 kilo base DNA sequence while Pfu Turbo polymerase has a 2.6% error rate on the same sequence. In our PCR amplification you use a DNA polymerase from the Archaea Pyrococcus furiosus (Pfu Turbo).
4.7 Applications of PCR
PCR is used to amplify DNA that is present only in a few copy numbers with several orders of magnitude. As a result PCR amplification generates millions of copies of the desired DNA sequence. PCR revolutionized molecular biology, and has a wealth of applications beyond basic science. A few of those applications are highlighted below.
- Specific DNA isolation: Using primers designed for the gene of interest a given DNA sequence can be amplified. This DNA can then be inserted into a cloning vector for further study or its sequence can be determined.
- DNA quantification (quantitative real-time PCR or qRT-PCR). This method estimates the amount of a given nucleic acid sequence present in the sample. Since theoretically each PCR cycle doubles the amount of the given nucleic acid the fewer amplification cycles it takes to generate a detectable amount of the DNA the higher its concentration was in the original sample. This means levels of gene expression can be determined. The method requires simultaneous amplification and detection (real-time). Primers amplify the nucleic acid of interest in the presence of a fluorescent dye that is specific to DNA. The number of cycles it takes to get detectable fluorescence intensities is quantified (CT number). This number is inversely proportional to the concentration of the target nucleic acid in the sample.
- Gene mapping. Using specific primers million –fold amplification of a given part of the genome can be achieved. That way, even if only a few copy numbers of the given DNA is available, using PCR sufficient amount of DNA can be generated for scientific studies. This trait of PCR resulted in a wealth of applications beyond basic science. DNA profiling (genetic fingerprinting) is a technique used by forensic scientists to identify an individual involved in a crime or determine parent-child relationship between individuals (paternity testing). Even though 99.9% of the human genome sequence is shared, there are sufficient differences between each human to allow identification. The most commonly used testing procedures today focus on short tandem repeats (STR). These regions of the human genome are very variable in sequence, this means it is extremely unlikely that individuals have similar sequences unless they are closely related. Only monozygotic twins, “identical twins”, have the same short tandem repeat sequences.
- PCR in diagnostics. Using specific primers sensitive to a given pathogen (bacteria or virus) the source of infection can be identified using PCR much quicker than by culturing samples. Likewise, mapping of specific parts of genome can reveal if an individual is prone to breast cancer or other diseases that are much more treatable if detected at an early stage.
PROCEDURES
Reagents and equipment needs are calculated per six student teams. There is ~20% excess included.
Equipment/glassware needed:
- PCR machine
- Three sets of micropipettes 20-100 μl, 2-20 μl and 1-10 μl
- 6 PCR tubes
- 6 centrifuge tubes
Reagents needed
- Pfu Turbo polymerase master mix (Stratagene); 130 μl total
- DpnI restriction enzyme (6 μl total)
- Primers to generate each mutant
- Plasmid DNA containing the ykkCD toxin sensor
To set up PCR reaction, carefully mix the following reagents, place tubes into the PCR machine and start protocol
- 20 μl 2X Pfu Turbo Mastermix
- 100 ng plasmid DNA (volume depends on DNA concentration)
- 1 μl 1 μM top primer
- 1 μl 1 μM bottom primer
- Water to 40 μl
The PCR machine is programmed as follows:
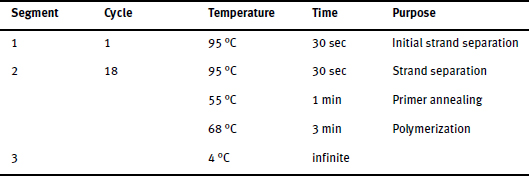
Remove template DNA (does not contain the mutation)
- Remove reactions from the PCR machine and briefly centrifuge them to ensure that all the reaction mixture is at the bottom of the tube.
- Add 1 μl DpnI restriction enzyme to each tube.
- Incubate reactions in a 37 °C water bath for 1 hr.
- Reactions may be stored at -20 °C until needed.
Notes to the instructor
The experiment in Chapter 4 is designed to perform site-directed mutagenesis on the Bacillus subtilis tetracycline sensor RNA ykkCD. The protocol with minimal modifications could be used to do site-directed mutagenesis on any nucleic acid. Usage of a high-fidelity DNA polymerase, such as Pfu Turbo, is essential for the success of the experiment, but alternative vendor or packaging may be used. Primers were ordered from Integrated DNA Technologies and reconstituted in 1 x TE (Tris-EDTA) at 1 μM concentration. Plasmid DNA containing the ykkCD sensor RNA is available upon request from the authors. Since this PCR reaction takes about two hours it is convenient to start the laboratory session by setting up the PCR reactions and giving pre-laboratory lecture while the reactions are running. This laboratory perfectly accommodates a long exam due to the minimal wet lab work and long reaction time. Depending on the time allocated for the lab session, PCR reactions may need to be removed by the teaching assistant or the instructor.
Prelab Questions for Site-Directed Mutagenesis
Define the following terms.
- Nucleotide

- Nucleic acid
- Reverse-complement

- Below is the bottom strand of a dsDNA. What is the sequence of top strand? Mark the 3’ and the 5' end of the top strand sequence.
3’ AAGTTCAAGGC 5’ - Calculate how you mix your PCR reaction if the concentration of your plasmid DNA is 275 ng/μl (use the Protocol section of your handout). Show your work.

Site-directed Mutagenesis
Lab Report Outline and Point Distribution
- Several sentences defining the goal/purpose of this experiment (2 pts.).
- Briefly describe the DNA polymerase reaction. Why is it thermodynamically favorable (about 5 sentences; 4 pts.)?
- Briefly describe Quickchange site-directed mutagenesis (5-10 sentences; 5 pts.).
- Outline the first cycle of a PCR reaction. Make sure you indicate the temperature that each cycle takes place. Briefly explain why you get significant amplification with PCR (4 pts.)?
- Outline the first cycle of Quickchange PCR. Explain briefly why you get less amplification with Quickchange then with regular PCR (5 pts.).
- PCR worksheet (30 pts.).
PCR Worksheet
DNA Structure:
1. (3 pts.) Draw the structure of pdApdCpdT. Label the 5´ and 3´ ends and circle each of the phosphodiester bonds in this small nucleic acid.
2. (3 pts.) Show the H-bonded pairing between adenine and its complementary base.
3. (3 pts.) A small double stranded DNA molecule is studied. Given the sequence of one DNA strand:
GGCTACATTCGGAA
write the sequence for the other DNA strand. (Remember, sequences are written from 5' to 3' direction).
Standard PCR:
4. (3 pts.) Why is PCR described as a “chain” reaction?
5. (3 pts.) Explain why two cycles of PCR are necessary before the desired DNA product is made.
6. (3 pts.) In PCR, a set amount of DNA polymerase is added at the beginning and must remain active through all subsequent cycles. This requirement severely limits which DNA polymerases may be used. Why is this so?
QuickChange DNA Synthesis:
7. (3 pts.) Site-directed mutagenesis allows scientists to specify exactly where DNA base changes (mutagenesis) will occur. How does the QuickChange process allow us to achieve “site-directed mutagenesis?”
8. (3 pts.) Standard PCR allows for “amplification” of the DNA product. Does QuickChange allow for the same type of “amplification?” Explain.
9. (3 pts.) Standard PCR requires that a primer bind at the beginning of the DNA template sequence. In contrast, a QuickChange primer does not have to bind at the beginning of the template but can bind anywhere along its sequence. How do you explain this difference?
10. (3 pts.) Standard PCR can be used to make many copies of a newly isolated gene. In contrast, QuickChange is only useful for making base changes in a well-studied DNA sequence. What is different about the QuickChange process that makes such a difference in how it is used?