
1.2: Identifying Conserved Elements in the Toxin Sensor and Designing Mutants to Test Whether They are Important for Function
- Page ID
- 18919

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)2.1 Learning Objectives
During this lab, you will use bioinformatics to learn how to find the DNA sequence of a given macromolecule and use this sequence to uncover evolutionary sequence conservation. You will use these data to identify conserved sequence segments (invariable blocks) in the ykkCD sensor RNA. During the second half of the lab you will identify conserved structural elements within the toxin sensor. These are elements where the sequence may have been altered during evolution, but the structure was retained. You will then use this information to design a mutant to see if a conserved sequence or structure is important for toxin recognition.
2.2 Mini Project Flowchart
The bolded blocks in the flowchart below highlight the role of the current experiment in the mini project.

2.3 Why is Sequence Conservation Important for Macromolecule Function, and How Do We Determine This?
As you learned earlier, the goal of the mini-project is to better understand the molecular basis of how the ykkCD sensor RNA recognizes the antibiotic tetracycline. You will determine which part of the sensor is essential for tetracycline recognition. The first step toward this goal is to identify segments of the toxin sensor (ykkCD riboswitch) that did not change throughout evolution. We call these segments invariable blocks. These elements are the most likely to play substantial roles in tetracycline recognition. You will subject these elements to site-directed mutagenesis and evaluate how these mutations affect tetracycline recognition by the sensor. Site-directed mutagenesis simply means that you will alter the sequence of the sensor in the bacterial DNA. If the mutation abolishes the ability of the sensor to recognize tetracycline, then the mutated part of the sensor is essential for tetracycline recognition (Fig. 2.1). You will conclude this if the KD (dissociation constant) value of a mutant sensor RNA – tetracycline complex is at least 10-fold larger (recall larger KD value means weaker binding) than that of the wild-type sensor tetracycline complex. If the KD value of the mutant sensor is within an order of magnitude of that of the wild-type sensor tetracycline complex you will conclude that the nucleotides altered were not essential for recognizing tetracycline.
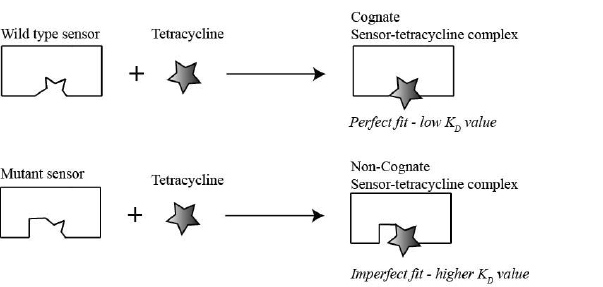
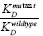 >10 (higher KD value) the nucleotide(s) mutated were important for tetracycline recognition. If <10 (low KD value) the nucleotides mutated were not important for tetracycline recognition.
>10 (higher KD value) the nucleotide(s) mutated were important for tetracycline recognition. If <10 (low KD value) the nucleotides mutated were not important for tetracycline recognition.2.4 Review of Nucleic Acid Properties
Before we identify conserved sequence elements and design mutants let us review a few things about nucleic acids. Nucleic acids are macromolecules that contain a chain of nucleotides connected by covalent bonds (phosphodiester bonds). Most nucleic acids contain all four nucleotides: adenine (A), thymine (T) or uracil (U), cytosine (C) and guanine (G). Our toxin sensor is an RNA molecule therefore it contains uracil instead of thymine, but since we perform our mutagenesis in the bacterial DNA we will see T instead of U in the sequence. The sequence of the nucleic acid is determined by the order in which the nucleobases follow each other in the macromolecule, and it determines the properties of the nucleic acid. For example even though the following two nucleic acids, ATCG and GTCA, contain the same bases, since the bases follow each other in different order, the properties of these two nucleic acids will be very different. Secondary structure, the interaction pattern between nucleobases, of a nucleic acid is easily predictable since an A always pairs with a T or a U using two hydrogen bonds and a C always pairs with a G using three hydrogen bonds. These pairs are referred to as Watson-Crick pairs or base pairs. Nucleic acid sequence comparisons and structure predictions are important applications of bioinformatics.
2.5 What is Bioinformatics?
During the last 15 years one of the greatest breakthroughs in biochemistry was the emergence of bioinformatics. Bioinformatics uses the power of computer science to analyze biology data and generate new information. This new field of life sciences has emerged, because (1) there has been an exponential increase in biological information due to genome sequencing, gene expression profiling (microarray data) and determination of macromolecular 3D structures, and (2) there has been a vast improvement in the access to computational power. The most common applications of bioinformatics in biochemistry are:
- Performing sequence comparisons to determine macromolecule function by comparing the sequence of a new macromolecule to that of macromolecules with known function.
- Generating sequence alignments to identify regions of a macromolecule that are conserved through evolution. These regions are likely to be important for function.
- Predicting secondary or tertiary structure of a macromolecule by comparing its sequence to macromolecules with known structure.
- Predicting and visualizing macromolecule-ligand interaction to determine how the macromolecule recognizes its specific target.
During this lab course we will familiarize ourselves with most of these applications.
Genome sequences are stored in the gene databank (GenBank) of the National Institute of Health (NIH). GenBank is the annotated collection of publicly available gene sequences. By the end of 2013 there was 169,331,407 sequences stored in the GenBank, which represents a 17-fold increase in data since the sequencing of the human genome in 2000 and an about 280,000-fold increase in data since the creation of GenBank in 1982. This database is available online free of charge to perform sequence comparisons, also called sequence alignments. There are two types of sequence alignments: global alignment and local alignment. Global sequence alignment aims to align as many characters as possible between the sequence of interest, the query, and sequences in the GenBank, the subject. The goal is to find a hit with high overall similarity. This method is slow, but is useful for example to uncover evolutionary relatedness between two species. In contrast, local sequence alignment focuses on stretches of high similarity, and thus, it compares discrete parts of the sequence of interest to sequences in the GenBank. Pairwise alignment aims to find the best way to match two sequences. Multiple sequence alignment compares the sequence of interest to many other sequences in the GenBank to find regions of the sequence that are conserved through evolution (Fig. 2.2).

The two major problems encountered when performing sequence alignments are: (1) Due to the vast amount of data available in the GenBank, it takes considerable time to perform a thorough comparison, and (2) sequences can be similar at random. To overcome these problems (a) sequences are divided into short segments (words) and these segments are simultaneously compared to sequences in the GenBank and (b) alignments are scored using a scoring function. The most commonly used alignment algorithm is Basic Local Sequence Alignment Tool or BLAST. BLAST uses a 7-15 nucleotide word size. Every time there is a nucleotide match between the query and the subject a +1 is added to the score. If there is nucleotide mismatch, a -2 is subtracted from the score. Often it is useful to introduce breaks (gap) into the alignment to generate a better overall match between two words. Introducing a gap results in a -3 penalty while extending a gap results in a -1 penalty. The alignment with the highest overall score is the best.
An example of how alignments are scored is seen below:
Query CCC Score = 1 match + 2 mismatches = (+1) + 2 x (-2) = -3
Subject GGCC
After shifting the query to the right:
Query CCC Score = 2 matches + 1 mismatch = 2 x (+1) + 1 x (-2) = 0
Subject GGCC
The second alignment has the highest score This means, it is the best.
An example of how the introduction of gaps improves alignments is seen below:
Query AGCAC Score = 2 matches + 2 mismatches = 2 x (+1) + 2 x (-2) = -2
Subject AGAC
After introducing a gap
Query AGCAC Score = 4 matches + gap = 4 x (+1) + 1 x (-3) = -1
Subject AG_AC
The alignment with the gap is a better match between the two sequences. BLAST divides the sequence of interest to words. Once the highest scoring arrangements are found the alignment is extended to find the best overall alignment. To account for similarity between sequences that takes place at random the Expected value or E-value is introduced. The E-value is a parameter that describes the number of random hits with a particular word size. Essentially the E-value represents the background noise (significance threshold) of an alignment. The closer the E-value is to zero the better is the alignment.
Secondary structure prediction of nucleic acids is straightforward, because secondary structure formation in nucleic acids follows the simple rule of Watson- Crick base pairing: A pairs with T or U using two H-bonds whereas G pairs with C using three H-bonds, thus G-C pairs are more stable than A-T pairs. Among the potential secondary structures the one with the lowest free energy is the most likely structure. The most popular algorithm to predict RNA structure is Mfold, where M stands for multiple, meaning that it generates more than one potential structure and fold stands for structure.
2.6 Identifying Conserved Sequence Elements (Invariable Blocks)
First you will use the sequence of the sensor from the model organism Bacillus subtilis to find toxin sensor sequences in different organisms. Then, you will compare the sequences of the toxin sensor in these different organisms using multiple sequence alignment to identify blocks in the sequence that did not change throughout evolution. Once you have identified the invariable blocks of the toxin sensor, you will choose one that you subject to mutagenesis. You will delete or insert nucleotides into the invariable block of your choosing, or modify its sequence.
2.7 Identifying Conserved Structural Elements
Besides invariable blocks, conserved structural elements may also serve as hot spots for recognition. These are regions in the molecule that may have different sequence, but form the same structure. For example, both the GGGG AAAA CCCC and the AAAA GGGG TTTT nucleic acids form a hairpin even though they have very different sequence. To identify conserved structural elements in the ykkCD toxin sensor first you will predict the secondary structure of the ykkCD sensor RNA using ykkCD sequences from different organisms. Then you will select structural elements that are present in each organism based on visual inspection of the predicted structures. To test if a conserved structural element is important for toxin recognition, you will design a mutant that significantly alters a conserved element, then design another mutant that restores the original structure (compensatory mutation). If the structural element was important for toxin recognition then significantly changing the structure would destroy recognition, but restoring the original structure, albeit with altered sequence, will restore the ability of the sensor to recognize its target toxin.
PROCEDURES
Identifying invariable blocks
- Go to http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- Click nucleotide blast.
- Paste the sequence below into the “Enter Query Sequence” window.
T G TA A AG T T T T C TAG G G T T C C G C AT G T C A AT T G AC AT G G AC
T G G T C C G AG AG A A A AC A C ATAC G C G TA A ATAG A AG C G C G T
ATGCACACGGAGGGAAAAAAGCCCGGGAGAG - Choose “others” under “Choose Search Set/Database” and “Somewhat similar
sequences” under “Program Selection”. - Hit BLAST.
You will see many sequences color-coded by the degree of similarity to the B. subtilis ykkCD RNA.
Find ykkCD sequences in the organisms below and paste them into your lab notebook!
Bacillus subtilis, Bacillus amyloliquefaciens, Bacillus licheniformis, Bacillus halodurans, Bacillus pumilus, Alkaliphilus oremlandii, Staphylococcus saprophyticus subsp, Symbiobacterium thermophilum - Perform multiple sequence alignment on the selected sequences by going to
http://bioinfo.genotoul.fr/multalin/multalin.html. - Each sequence should be entered in a different row with designation preceding the sequence as follows:
>Sequence name (for example B.subtilis)
sequence (for example Tgtaaagt……)
>Sequence name
sequence
etc. - Hit “Start MultAlin”.
- Nucleotides colored in red represent invariable groups.
Identifying conserved structural element
- Go to mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form.
- Paste the RNA sequences below into the program window to predict their structure.
B subtilis sequence
UGUAAAGUUUUCUAGGGUUCCGCAUGUCAAUUGACAUGGACUGGUCCGAGAGAAAACACAUACGCGUAAAUAGAAGCGCGUAUGCACACGGAGGGAAAAAAGCCCGGGAGAG
Staphylococcus saprophyticus sequence
AAAACUGGCUUCUAGGGUUCCGGUCCCGCUCCUGUGGUGGGACGGCUGGUCCGAGAGAAGCA.GCCG..GUCCGACAGCAGGGCCGGUCACACGGCGGGAGAAAAGCCCGGGAGAG
Gloeobacter violaceus
AAUAAAGCUUUCUAGGGUUCCGCAAGGUGAUUACUUUGGUCUG.GUCCGAGAGAAAGCCACAUAUUUUUAUGUGACACGGAAGGAUAAAAGCCUGGGAGAU - Choose “Fold RNA”.
- To view predicted structures go to “View Individual Structures”. Select jpeg
format.
Mutant design (design three mutants)
- Propose a mutant that changes a conserved sequence element of the toxin sensor (mutant 1).
- Predict the structure of this mutant using Mfold.
- If the mutant changed the structure of the sensor significantly (predicted structure is completely different from the original one) choose a different mutant until you find one that changed a conserved sequence without significantly changing the overall structure.
- Design mutants to test if a conserved structural element is important for toxin recognition by following the outline below.
- – Propose a mutant that significantly alters or eliminates a conserved structural element (mutant 2).
- – Design a compensatory mutant to restore the original structure (mutant 3).
Notes to the instructor
The experiment in Chapter 2 is designed to identify sequence conservation in the B subtilis tetracycline sensor RNA ykkCD. The same protocol with minimal modifications could be used to identify sequence conservation in any nucleic acid (regulatory RNA, promoter or ribozyme). Two students per computer works well to maximize peer interaction while still making sure that each student has a chance to intellectually contribute to the assignments. Tablets or smart phones may also be used to complete each task. This means, this experiment may be used as an assignment in a lecture course. The websites listed above are free to use and have access to the latest entries in the nucleic acid sequence data bank, but the following websites can also be used as alternates if deemed necessary:
http://embnet.vital-it.ch/software/ClustalW.html (CLUSALW to perform sequence alignments)
rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi (Vienna Package to predict nucleic acid secondary structure)
BLAST Prelab
Nucleic acids review
- Nucleic acids are polymers of_____________________________. ( / 1 pt.)
- Nucleic acid polymers (strands) go from ___ to 3' direction. ( / 1pt.)
- GC pairs have _________H-bonds while AT pairs have _______H-bonds, therefore AT pairs are ___________ (choose more or less) stable than GC pairs. ( / 3 pts.)
- Draw the structure of ATP. ( / 1pt.)
- Indicate sugar, phosphate and the nucleobase on the nucleotide below. ( /3 pts.)
- Write the complementary sequence for the following nucleotide. ( / 1 pt.)
5’ GCCGATA 3’
3’ 5’
Questions regarding “Identifying invariable blocks in the toxin sensor”
- Circle “invariable blocks” in the following sequence alignment. ( /2pts.)

- Circle conserved structural elements (structures that all of these RNAs have in common). ( / 2pts.)
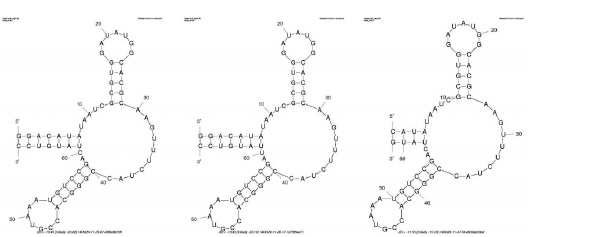
Identifying Invariable Blocks in the Toxin Sensor
Lab Report Outline and Point Distribution
- Define the goal of the experiment (3 pts.).
- Copy of multiple sequence alignment (3 pts.).
- Circle each invariable block (3 pts.).
- Secondary structure predictions for Bacillus subtilis, Staphlococcus saprophyticus and Gloebacter violaceus ykkCD sensor RNAs. (3 pts. each = 9 pts. total).
- Circle the conserved structural elements for the three structural predictions performed (3 pts.).
- Show structural prediction for mutant 1 (2 pts.).
- Comment on why you believe that your mutation caused these specific structural changes (5 pts.).
- Show the structural prediction for mutant 2 and mutant 3 (4 pts.).
- Comment on why you believe that mutant 3 restored the original structure (5 pts.).
- Based on your experience with mutants 1 and 2 explain why changing structure and sequence at the same time makes it hard to interpret the functional effect of a mutation (aka what was the reason for designing mutant 3) (7 pts.).
- Choose a mutant (from the three designed here) that you would like to test experimentally. Provide a brief explanation for why you chose this mutation (6 pts.).
BLAst Problem Set
Define or describe the following terms as used in BLAST:
- Word
- E (Expect or Expectation value)
- Gap
- Max Score
Find and report the following:
- The number of identities in the alignment between the query and the subject, Cyanothece sp. PCC7425
- The percent of coverage between the query and the subject, Sulfuricurvum kujiense DSM16944
- The number of gaps in the alignment between the query and the subject, Clostridium kluvyeri DSM555
- The Expect value in the alignment between the query and the subject, Exiguobacterium sibiricum 255-15, complete genome.
- The percent of maximum identity between the query and the subject, Geobacillus sp Y412MC52
Find and interpret the following:
- Find the E value for the alignment between the query and the subject, Bacillus subtilis subsp. spizizenii str. W23, complete genome. How do you specifically interpret this number? (What does this number mean?)
- Find the E value for the alignment between the query and the subject, Pseudomonas stutzeri DSM 4166. How do you specifically interpret this number? (What does this number mean?)
- For the alignment between the query and the subject, Paenibacillus sp. JDR-2, complete genome, there are fourteen gaps reported yet only five gap openings are shown in the alignment. Is this a mistake? Briefly explain.
General BLAST interpretation:
- The search using megablast gave far fewer matches than the search using blastn. Check the Algorithm Parameters for both searches. Could the difference in word length explain the difference in matches? Briefly explain.
- The alignment with the subject, Clostridium cellulovorans 743B complete genome, has a smaller % identity than the subject, Lysinibacillus sphaericus C3-41, complete genome. Yet, these two subjects have the same maximum score. Briefly explain why this is the case.
Protein Properties Worksheet
In the past 10 years, one of the greatest improvements in biochemistry took place in the field of bioinformatics. Search engines and prediction tools enable us to easily learn a lot about a protein or RNA of interest before we actually start working on a project. The availability of these online tools makes it much easier to tailor a project and assess its feasibility. This problem set introduces you to a few easy-to- use prediction tools. You will learn how to calculate the molecular weight, isoelectric point or amino acid composition of a protein, how to perform sequence alignments to assess sequence conservation, how to predict secondary and tertiary structure.
Follow the step-by-step instructions below and answer all questions
You will use the sequence of the ykkCD multidrug-resistance efflux pump. Efflux pumps are an important part of bacterial defense against antibiotics: they pump antibiotics out of the bacterial cell thereby rendering them ineffective in treating bacterial infections. The RNA sensor (ykkCD riboswitch) you perform mutagenesis on in the biochemistry lab turns on production of the ykkCD efflux pump.
The ykkCD efflux pump is a heterodimer pump meaning that it is made out of two different proteins: ykkC and ykkD. In the first part of the exercise you will perform a series of predictions using these sequences to get an idea about the physico-chemical and structural properties of this pump.
The sequences of the ykkC and ykkD pumps are listed below:
>ykkC
MKWGLVVLAAVFEVVWVIGLKHADSALTWSGTAIGIIFSFYLLMKATHSLPVGTVYAVF TGLGTAGTVLSEIVLFHEPVGWPKLLLIGVLLIGVIGLKLVTQDETEEKGGEA
>ykkD
MLHWISLLCAGCLEMAGVALMNQYAKEKSVKWVLLIIVGFAASFSLLSYAMETTPMG TAYAVWTGIGTAGGALIGILFYKEQKDAKRIFFIALILCSAVGLKILS
- Generate a sequence alignment between the two pump monomers (C and D) using this link: http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html. Make sure you copy/paste both sequences into the program as it is shown above (names and > included).
- a) Paste the sequence alignment!
- b) How similar are these two sequences (% conservation)?
- c) Is there any trend in the types of amino acids that appear conserved (apolar/polar/charged)?
- d) Based on “c” what prediction would you make about the structural similarity of these two proteins (similar or not and why)?
- Choose the ykkC or the ykkD pump to perform a series of predictions using the link below http://www.expasy.ch/tools/protparam.html; pI (isoelectric point), MW and amino acid composition. You may use the ykkC or the ykkD pump sequence for this excersize!
- a) List pI (isoelectric point) and molecular weight (MW) of this protein.
- b) What are the three most common amino acids in this protein?
- c) What can you tell about the general physical properties (polar/apolar; acidic/basic) of this protein? Would you expect this protein to be water soluble?
- Predict the secondary structure of the ykkC or the ykkD pump by using: http://www.compbio.dundee.ac.uk/www-jpred/
- a) Paste the result of the secondary structure prediction into your report!
- b) What is the major secondary structural motif in this protein?
- c) Based on your secondary structure prediction which amino acids are prone to form a-helix?
- Using the link below predict membrane topology of the pump used in “3”. http://phobius.sbc.su.se/. Answer the questions below
- a) Paste the prediction here.
- b) Based on the secondary structure prediction in “3” and the previous topology prediction sketch how the pump is inserted into the membrane.
- Tertiary structure prediction: Below is the sequence of the MexA multidrug resistance efflux pump from the pathogenic organism Pseudomonas aeruginosa. This organism mostly affects patients with compromised immune system. It is the main culprit behind infections caused by medical implants (like catheter).
Perform tertiary structure prediction on this protein using the link and the sequence below!
swissmodel.expasy.org/workspace/index.php?func=modelling_simple1
Warning! Performing this modeling will take a while (10-20min). Do not use the back button on your browser; just wait patiently until it is done.
>MexA
MQRTPAMRVLVPALLVAISALSGCGKSEAPPPAQTPEVGIVTLEAQTVTLNTEL PGRTNAFRIAEVRPQVNGIILKRLFKEGSDVKAGQQLYQIDPATYEADYQS AQANLASTQEQAQRYKLLVADQAVSKQQYADANAAYLQSKAAVEQARINLRY TKVLSPISGRIGRSAVTEGALVTNGQANAMATVQQLDPIYVDVTQPSTAL LRLRRELASGQLERAGDNAAKVSLKLEDGSQYPLEGRLEFSEVSVDEGTGSVTIRAV FPNPNNELLPGMFVHAQLQEGVKQKAILAPQQGVTRDLKGQATALVVNAQNKVEL RVIKADRVIGDKWLVTEGLNAGDKIITEGLQFVQPGVEVKTVPAKNVASAQKADAA PAKTDSKG
a.Paste the picture of the resulting structural model into your report!
b.What can you tell about the distribution of polar and apolar amino acids in a membrane protein as compared to a water soluble protein?