
Review: Organic Chemistry
- Page ID
- 4599

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Breaking C-X bonds
Many of the organic reactions involved in metabolism involve making and breaking bonds to carbon. There are 3 ways to break a bond to a C-X bond, producing either a carbocation, carbanion, or free radical intermediate, all of the which are unstable and reactive. Both the carbocation and free radical are electron deficient, and the carbanion, although not electron deficienct, has a negative on C, an atom which has a relatively low electronegativity.
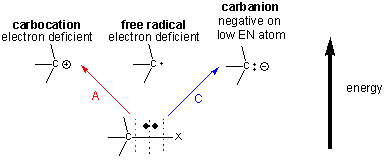
Preferential Stabilization of Reactants, Intermediates, and Products
These unstable intermediates are higher in energy than the reactants, and hence the transition state, which is even higher in energy than the intermediates, must have a structure which resembles the intermediates more than the reactants. (i.e. for the charged carbanion and carbocation intermediates, there is a developing charge in the transition state.
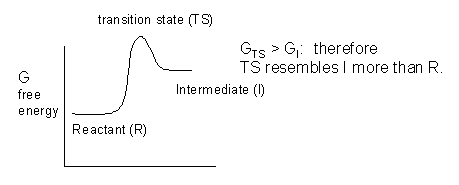
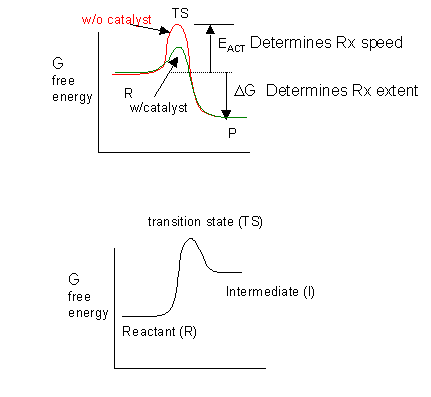
The thermodynamics of the reactions is determined by the change in free energy between the intermediates and the reactants, while the kinetics of the reaction is determined by the difference in free energy between the transition states and the reactants. A catalyst lowers the energy of the transition state without affecting the energies of the reactants or intermediates (assuming that these are free and not bound to the catalyst.
A comparison of the thermodynamic reactivity of molecules of similar structures can be made by determining the relative stability of the reactants and products from structural considerations. Consider two reactants, R1 and R2, which produce products P1 and P2, respectively. Any structural features that preferentially stabilizes R2 compared to R1, or P2 compared to P1, but doesn't stabiiize R2 and P2 to the same extent, will lead to a greater driving force for R2 --> P2 compared to R1. This is shown graphically below.
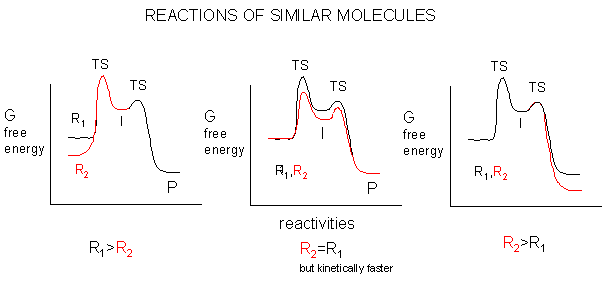
Mechanisms which lead to the stabilization of a reactant, intermediate, or product include resonance and inductive effects (electron release or withdrawing).
- An example of how the comparative acidity of two similar molecules can be determined through a comparison of their structures is shown below for acetic acid and ethanol.
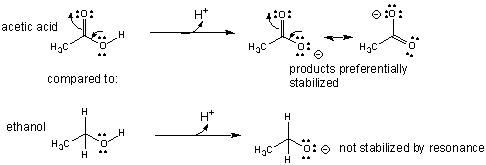
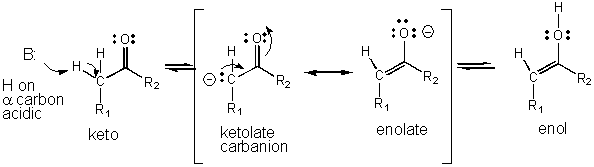
- An example of how an intermediate can be stabilized through resonance is shown below for the keto - enol tautomerization reaction which is favored in the direction of the keto form, a a weaker acid than the enol.
- An example of how the inductive effect (electron release and withdrawal) stabilize/destabilize carbocations and cations is shown below.
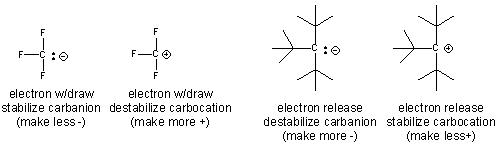
- Electron withdrawing by the F's in the negatively charged conjugate base of trifluoracetic acid helps explains its lower pka compared to acetic acid.
- stabilization of the tertiary carbocation below helps explain the preferential formation of the tertiary alcohol
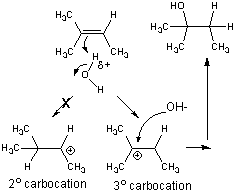
Oxidation of Organic Molecules
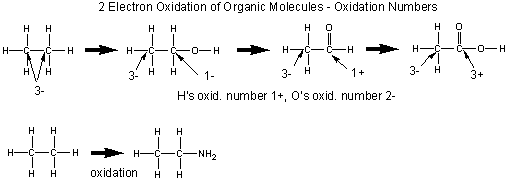
Organic molecules are usually oxidized in two electron steps. Two methods can be used to determined if a C atom in an organic molecule has been oxidized.
- If the number of bonds from the C to oxygen increase, or the number of bonds to H decrease, the C is oxidized, More generally, if the number of bonds from C to a more electronegative atom increases, or the number of binds from C to a less electronegative atom decrease, the carbon is oxidized
- A more powerful method involves determining the oxidation number of the carbon atoms in the reactant and product. If the oxidation number becomes more positive, the C is oxidized.
The general rules for determining oxidation numbers of the atoms in a molecule are:
- O is generally 2-
- H is usually 1+
- in molecules consisting of one type of atom, (like O2) - i.e. a polyatomic element, the atoms have an oxidation number of 0.
- the sum of the oxidation numbers of the atoms in a molecule equals the net charge on the molecule or ion.
In general, the oxidation number can be calculated as follows:
- assign all nonbonded electrons of an atom to that atom
- assign all bonded electrons to the more electronegative atom of the two atoms bonded
- assign one electron of a bond to each atom if the two atoms are identical.
- sum up the assigned electrons from 1-3. Subtract this number from the total number of electrons usually present in the outer shell of the atom (the group number). The result is the oxidation number.
An illustration of the sequential two step oxidation of ethane to acetic acid and assigned oxidation numbers.
Reaction of Carbonyls: Aldehydes and Ketones
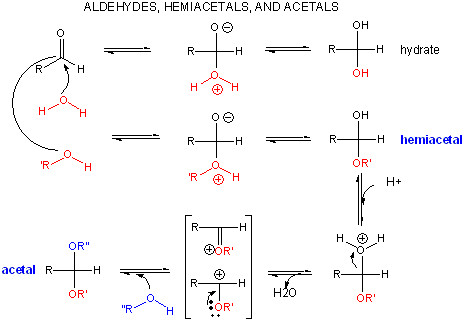
When water reacts with an aldehyde in a nucleophilic addition reaction, a 1,1 diol, or a geminal diol results. This reacts can be catalyzed by base, which acts as the nucleophile (it's a stronger nucleophile than water) and adds to the carbonyl C. OH- is regenerated when the alkoxide produced abstracts a proton from water, regenerating OH-.
When an alcohol adds to an aldehyde or ketone, a hemiacetal or hemiketal, respectively, is formed. In the presence of an acid catalyst, the acid protonates the carbonyl oxygen, making the carbonyl more electrophilic. After the alcohol adds and forms the hemiacetal or hemketal, the acid can protonate the OH group, leading to its expulsion as water in an acid-catalyzed elimination. The carbocation or resonant-form oxonium ion can react with another ROH to form an acetal or ketal.
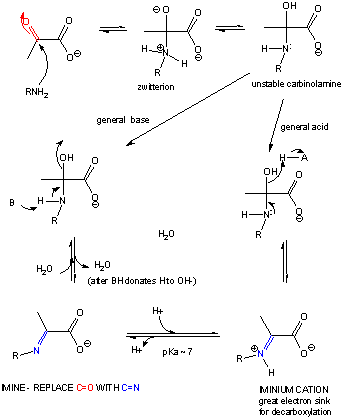
If the nucleophile is an amine, an addition can occur, followed by an elimination to form an imine or Schiff base..
An acetal or ketal are really geminal ethers (as water addition to aldehydes or ketones produced geminal diols). As with other ethers, these geminal ethers are stable to base, and are hence often used as protecting groups to keep aldehydes and ketones from undesired reactions in basic solution. Acetal formation is favored by excess anhydrous alcohol in acetic condition, while acetal breakdown is accelerated by high concentrations of water and the presence of an acid catalyst.
Why are ethers and hence acetals/ketals resistant to bases? They are resistant to nucleophilic attack, such as by base, since the expelled group (alkoxide) is unstable. (Epoxides, in contrast, will react with OH- nucleophiles since the epoxide ring is strained and of high energy.). Ethers can react with acids however, which protonate the ether O to form an oxonium ion. Nucleophilic attack (such as by Br-) on an adjacent C can occur (SN2), with electrons flowing to the protonated oxonium ion (a great electron sink) as it departs.
Reaction of Carboxylic Acid Derivatives
Carboxylic acids undergo nucleophilic substitution reactions, assisted by the fact that compared to aldehydes and ketones, they have good leaving groups. With the substitution reaction, the stability of the double bond in the carbonyl is retained. Two things control the reactivity of these derivatives: the stability of the reactants and the stability of the products.
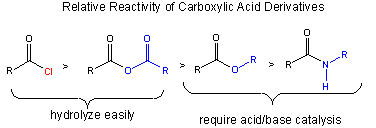
The stability of the reactants: A reactant is less reactive if stabilized by resonance. The figure below shows that the order of increased reactivity of reactants is:
amide < ester < anhydride < acid chloride
The nonbonded electron pair on N of the amide, being less electronegative than O, can delocalize and form a resonant structure with a double bond with the carbonyl C more readily than the O in the ester. An electron pair on the bridging O in the anhydride, since its ability to from a double bond in a resonance structure is split between the two carbonyls C is less effective in stabiliziing either side than in the. (This is called competing resonances.) The reactant less stabilized by resonance is the acid chloride, since a nonbonded pair of electrons on the larger chlorine molecule can't delocalize as readily given C-Cl bond distance.
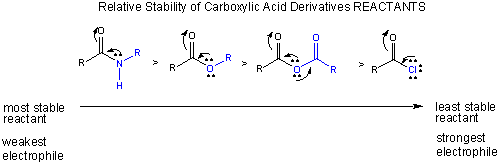
Notice this order of decreased stability based on resonance stabilization is also the order of increased electrophilicity of the carbonyl C (which is most electrophilic in the absence of electron delocalization from the adjacent N, O, or Cl.
The stability of the products: If the deprotonated leaving group is considered as one of the products (whch differentiates the different reactions), then the order of decreased stability of products is:
Cl- > RCOO- > RO- > RHN-.
(Note: the pKa of ROH = 16, R2NH = 40)
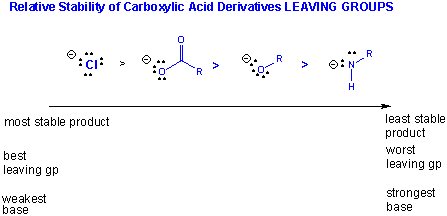
More on the Thermodynamics of carbonyl reactivity:
What really determines the stability of products compared to reactants is the strength of bonds made and broken during the reaction.
- In nucleophilic addition to aldehydes and ketones, the strength of the bond to the nucleophile must be greater than the strength of the pi bond broken in the carbonyl. A C-Cl bond strength is 81 kcal/mol compared to a pi C-O bond strength of 93 kcal/mol. Hence a Cl- is not likely to add to a carbonyl C. Consider the hydration of formaldehyde (carbonyl with 2 H's), acetaldehyde (with 1 H and 1 methyl group, and acetone (with 2 methyl groups). The ΔGo for hydration of these is -19, -1, and +15 kcal/mol, respectively, showing that increased electron release toward the carbonyl C, which makes it less electrophilic and more stable, decreases the reactivity of the carbonyl.
- In nucleophilic substitution, the leaving group (anion) must be more stable than the nucleophile.
Kinetics of Reactivity of Carbonyls:
The relative kinetic reactivity of various carbonyl's toward nucleophiles follows the order of electrophilicity of the C. (i.e the extent of the positive charge on the carbonyl C.) The slow step in a nucleophilic attack is breaking the pi carbonyl bond. If the reactant is stabilized by resonance in ways that reduces the electrophicity of the carbonyl C, the reaction is slowed.
Nucleophiilicity really is a measure of the affinity of an atom or ion on an electrophilic C which is similar to basicity which is a measure of the affinity of an atom or ion for a proton. Halides are not good nucleophiles for reactions with acid derivatives since the halide (like Cl-) is a better leaving group than the actual leaving group.
Making C-C Bonds
Metabolism can be divided into catabolic (breaking down) and anabolic (synthetic) reactions. To obtain energy, sugars and fatty acids are converted to carbon dixoide. Hence C-C bonds must be broken. In contrast, C-C bonds must be synthesized in photosynthesis. In all reactions, electrons from bond broken flow to atoms where bonds will be made. Flow is from a source (a pair of electrons possibly with a negative charge) to a sink (a slightly or fully positive atom). Here's a couple ways to make a C-C bond:
- react two carbon centered radicals (free radical mechanisms are uncommon biologically)
- react a carbocation with a carbanion.
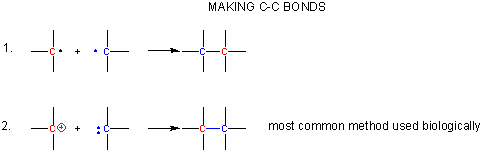
A carbocation is unstable unless incorporated into a molecule in which it is stable, so instead of using them the carbonyl C is used as the electrophilic carbon. (Instability of carbocations is reflected in their propensity to rearrangement.) Taking into account the resonance form of the C-O carbonyl bond in with a positive on C and a negative on O, the net charge on the carbonyl is about +0.5. A stabalized carbanion, often stabilized as a enolate resonant form, is used as the negatively charged carbon.
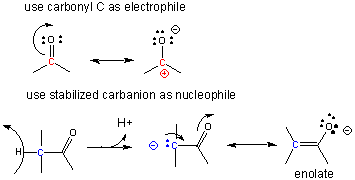
One method of making a C-C bond is an aldol condensation, in which a carbanion formed the deprotonation of a C-H alpha to a carbonyl (which is stabilized by the enolate resonance form) acts as a nucleophile which addes to a carbonyl C in an aldehyde or ketone.
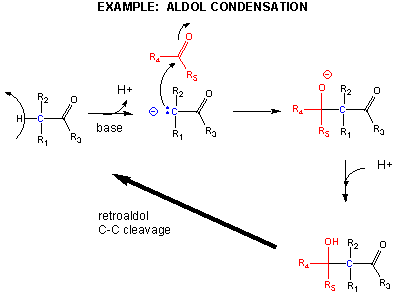
In another C-C bond synthesis reaction, a Claisen Condensation, a carbanion formed the deprotonation of a C-H alpha to a carbonyl (which is stabilized by the enolate resonance form) acts as a nucleophile which substitutes at a carbonyl C in an ester,
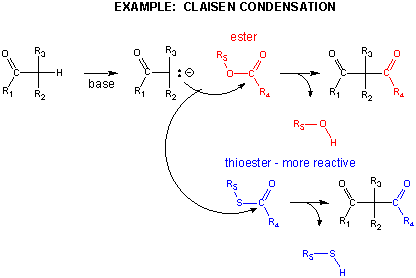
Breaking C-C Bonds
In addition to a retroaldol condensation, a common method to break a C-C is through a decarboxylation reaction at a beta-keto acid. Notice that the analogous reaction at an alpha keto acid is unlikely since the electrons from the C-C bond that is cleaved have no "sink" to which to flow.
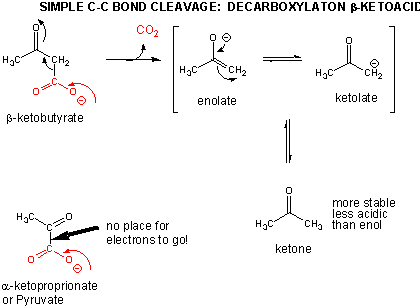
References
- Sole et al. Fleeting Molecules Extend Their Stay. (about carbenes) Science. 292, pg 1846, 1901 (2001)
- On the Threshold of Stability (Carbene Chemistry). Nature 412, pg 598 (2001)
- Wang et al. Construction Principles of "Hyparenes": Families of Molecules with Planar Pentacoordinate Carbons. Science. 292. pg 2465 (2001)
- Superacids: it's a lot about anions. Science. 289. pg 72, 101 (2000)
- An end to the protection racket. (use peptide catalyst and avoid using protecting groups). Nature. 414, pg 703 (2001)