
2.14: Session 15
- Page ID
- 26437
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)SESSION 15
Today you will
- Analyze your sequencing data from the site-directed mutagenesis
- Use the PyMol structure viewing program to view wild type and mutant Abl crystal structures, and complete the structure viewing worksheet.
1.) Discuss the results of the activity and inhibition assays as a class.
2.) Analyze your sequencing data for the site-directed mutagenesis using DNA strider or another DNA sequencing program. Print out a copy of the DNA analysis for your final report.
3.) Use PyMol to analyze crystal structures of Abl binding to inhibitors and complete the structure viewing worksheet.
STRUCTURE VIEWING
For this exercise you will use the structure-viewing program PyMol to analyze crystal structures from the protein data base (PDB) of the Abl kinase domain bound to various small-molecule inhibitors.
You will view the following crystal structures:
- PDB entry 1IEP: wt Abl kinase domain bound to Gleevec
- PDB entry 2GQG: wt Abl kinase domain bound to Dasatinib
- Optional… PDB entry 2F4J: the (Gleevec-resitant) H396P mutant of the Abl kinase domain bound to the kinase inhibitor VX-680
You will need to download the PDB file for each structure. Go to www.pdb.org, and in the top search bar type the 4-character ascension code (ie. 1IEP) for the desired structure.
Once you are on structure page, on the left hand column under “Download Files” choose “PDB text” and save the file somewhere you will be able to find it later. Do this for all three structures.
Please keep the following points in mind as you view these structures
- The wt structures 1IEP and 2GQG each include two independent molecules (meaning that two kinase domains are in each crystal, and that the two are not interacting) in addition to the small molecule inhibitors. In viewing these structures, you will select a single copy of each kinase domain (“chain a” to observe.
- The numbering of the Abl structures is identical to that in your 5.36 lab manual.
Please type or neatly write out answers to the following questions on a separate sheet of paper (complete sentences are not required). This should be turned in with our final lab report. There are a total of 22 questions.
Secondary Structure
First, we will look at the overall secondary structure of the Abl kinase domain. For this part of the exercise you should use the 1IEP structure
In the File menu, choose Open and chose the PBD file you downloaded previously. This will open the molecule in the viewer window.
Here are some instructions to help you work with this structure in PyMol:
- Under the Display menu choose Sequence, this will display the sequence at the top of the viewer window. You can choose residues in the sequence by clicking on them here, and they will be highlighted in the viewer window.
- In the command line, type select chain a, Chain a. A tab called "Chain a" will show up in the right hand column of the viewer window. Selecting an individual chain will allow you to view a single copy of the kinase domain, rather than view both copies that are present in the 1IEP structure.
Each selection in the right hand column has five menus: Actions, Show, Hide, Label, and color (A,S,H,L,C)
- Next to 1IEP click on H (for hide), choose everything. This should make the whole structure disappear.
- Next to “Chain A” click on the S (for show), and choose cartoon. Now the cartoon ribbon diagram of just chain A should appear on your screen.
- To make manipulating this chain easier, in the “Chain A” menu click on A (for actions), then center. To help orient yourself, you may want to turn the molecule around until it is in the orientation of the kinase domains in the Lecture 4 notes.
Other cool things you can do:
- Color by secondary structure: under C choose ss, choose either color scheme. This can help you locate secondary structure elements.
- Color as a rainbow: under C choose spectrum, then rainbow. This will color the structure from blue at the N-terminus to red at the C-terminus, the sequence at the top will also change color accordingly.
Questions:
1) What type of secondary structure is most prominent in Abl?
2) What type of secondary structure is most prominent in the N-lobe (residues 225-350) of Abl?
3) What type of secondary structure is most prominent in the C-lobe (residues 354-498) of Abl?
4) The protein contains a five-stranded β-sheet. Is this sheet located at the C or N terminus of the kinase domain?
5) What residues comprise this beta-sheet? For this answer do not include the helix that is betweent eh strands, so you should list two ranges. (for example "S229-G250 and L340-A350" would in the correct form- although this is not the correct answer). It may be helpful to zoom in on your structure to answer this question.
One can also evaluate the type of residue present in different parts of a protein. In this case it may be easier to look at the structure as a stick model and not a riboon model. In the "Chain A" menu show (S) sticks and hide (H)cartoon.
To color the structure by residue type in the command line type color white, resn val+trp+..... (type in three letter codes for each residue of the type you are coloring)
Using the command above, color all of the hydrophobic residues white (leave out glycine). Remember to include the aromatic residues for a total of 9 hydrophobic amino acids.
Questions:
6) Where are most of the hydrophobic residues located, on the inside or the outside of the protein? (You may want to switch back and forth from viewing the sticks to the cartoon from to get a better view of this.)
7) Where are most of the polar residues located?
8) Does this make sense in terms of protein folding? Explain.
Molecular Details
Now we will look more closely at specific regions and residues in the !IEP structure to understand the molecular basis for inhibition of Abl by Gleevec and how mutations can confer Gleevec resistance.
- If your structure is currently in the stick form, swithc back to the carton form. In the "Chain A" menu hide (H)sticks and show (S)cartoon.
- To reset the color of your structure in the Chain A tab choose color, by element and choose the first coloring scheme, then choose color, by chain and choose the first coloring scheme.
Binding of Gleevec
Now let's look more closely at the bdinging of Gleevec to the Abl kinase domain.
- In the command line type: select Gleevec, organic. Press enter. A new tab will appear on the right hand menu. This i the menu that controls the Gleevec molecule.
- For Gleevec, change the color to a different color from Abl. For example choose color and select orange. Next go to color, by element and choose the first coloring scheme.
Although Bcr-Abl is constitutively active, it may adopt an activated state or one of many inactivated states. Consider Gleevec binding in relation to the activation loop (A loop) of the Abl kinase domain. The A loop is comprised of Abl residues 381-402.
- In the command line type: select Aloop, resi 381-402. Press enter. A new tab will appear on the right hand menu to control the A loop.
- Highlight the A loop with a new color. Under the “Aloop” tab, select color and then magenta
- Select the conserved DFG (Asp-Phe-Gly) motif within the A loop by typing: select DFG, resi 381-383. Press Enter.
- In the “DFG” menu on the right of the screen, select show (S) as sticks.
Questions:
9) When bound to Gleevec, is the A loop of the Abl kinase domain in the open (extended) or closed conformation? Very briefly explain your answer.
10) When bound to Gleevec, is Abl kinase domain in the active or inactive form. Very briefly explain your answer.
Hydrogen bonds in the active site
Gleevec binds in the ATP-binding pocket, the active site, of Abl. One important thing to note is that the resolution of an X-ray crystal structure is not high enough to see hydrogen atoms, so they are not included in the model. To assign hydrogen bonding interactions which are important in substrate, you must read beyond what the structure can tell you and use your chemical knowledge.
It may be easier to see molecular interactions if you restrict the amount of atoms you see on the screen.
- In the command line type: select bindingpo select all atoms within 8 angstroms of the Gleevec molecule and make a menu for it in the right hand menu column called “bindingpocket”. Note: do not use spaces in your selection names. Also, PyMol is case sensitive so if you named the selection “Gleevec” it will not understand “gleevec”.
- In the Chain A menu choose H, everything.
- In the bindingpocket menu choose S, sticks or lines (sticks are thicker than lines).
To measure distances between bonds, click on the Wizard menu (at the top of the command line window) and choose measurement. In the window it will prompt you to click on the first atom. Follow the prompts to measure distances as needed to answer the following questions.
In looking for hydrogen bonds, make sure the atoms you are measuring are atoms that could be hydrogen bonding partners, and remember that reasonable H-bonding distances are 2.4 - 3.0Å
If your screen is getting cluttered with distance values, click "Delete all Measurements" in the right hand menu. When you are done measuring distances, click Done.
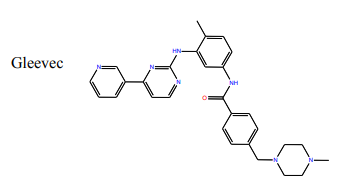
Questions:
11) What group on the structure of Gleevec above could participate in hydrogen bonds? (Draw Gleevec and indicate teh groups by circling).
12) Looking at the Abl structure, name the residues that appear to hydrogen bonds to Gleevec and give the distance between the atoms. Draw these on your Gleevec structure. Hint: there are 4 possible hydrogen bonds within 3.0 Å.
Note that only new hydrogen bonds make inhibitor binding energetically more favorable. H-bonds with an inhibitor that result from breaking H-bonds among residues within the binding pocket or with coordinated water do not significantly stabilize inhibitor binding.
13) Name two other interactions that may be stabilizing the inhibitor in the active site.
Let's now consider the most common mutation site found in cases of Gleevec-resistant CML. site 315.
14) What amino acid is at this site, and what sort of interaction does this particular residue have with the inhibitor?
15) What might you predict would happen if this residue were mutated to Asn or Ile?
16) What if this residue were mutated to Ala?
Now consider residue 315 in the structure of Abl complexed with Dasatinib, the other inhibitor you tested in the session 13/14 kinase activity assays. For this we will use the structure with PDB ID: 2GQG. In this structure there are again two molecules in the file, so you should select a single chain to look at.
Close the 1IEP structure and open 2GQG in the viewer window.
- Under the Display menu choose Sequence.
- Type select chain a, Chain a. Next to "2GQG" click on H, and choose everything. Next to "Chain A" click on the S and choose cartoon.
- As with the Gleevec-bound structure, to make manipulating this chain easier, click on A, then center in the "Chain A"menu.
Once you have opened the structure, consider the inhibitor-binding site. This is the same site Gleevec binds to. but Dasatinib binds ina different way.
Select the inhibitor (Dasatinib) by typing select Dasatinib, organic and pressing enter.
Change its color so that you can distinguish it from the protein. (See the directions above for Gleevec if you do not remember how to do this.)
As a reference, here is a structure of the Dasatinib:
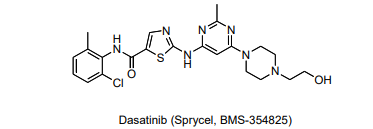
Look at residue 315 of Abl. Highlight this residue on the protein by clicking on the corresponding “T” in the sequence display at the top of the screen. The chosen residue (315) will be highlighted, and in the right hand menu there will be a tab labeled “sele”. This tab controls whatever you have selected in the viewer window. Use this tab to display residue 315 in stick form and change the color. Refer to the detailed instructions from above to manipulate the residue as needed to answer the following questions.
17) Is residue 315 interacting with Dasatinib? If so, describe the interaction.
18) Would you expect Dasatinib to effectively inhibit an Abl mutant with residue 315 mutated to an Ile? Very briefly explain why or why not.
Conformational Considerations
Now consider Dasatinib binding in relation to the activation loop of Abl kinase.
- As you did with the previous (Gleevec-bound) structure, select the activation loop by typing select Aloop, resi 381-402 and pressing enter.
- Next to the "Aloop" tab, select color and then magenta.
- Select the conserved DFG (Asp-Phe-Gly) motif within the A loop by typing: Select DFG, resi 381-383. Press enter.
- In the "DFG" menu on the right of the screen, select show (S) as sticks.
19) When bound to Dasatinib, is the A loop of the Abl kinase domain in the open (extended) or closed conformation? Very briefly explain your answer.
As you may recall from Lecture #4, Tyr 393 within the activation loop mimics the target tyrosine (to be phosphorylated) on the peptide or protein substrate, and it binds in the substrate-binding site of Abl. When Tyr 393 is phosphorylated, it can no longer bind in this site. Tyr393 is often phosphorylated in the active form of a kinase, but is never phosphorylated in the inactive form.
Find residue Tyr393 in the Dasatinib-bound Abl structure. Note the location of this residue in this structure. (Residue 393 is listed as “PTR” instead of “Y”. “PTR” stands for “phosphoryl tyrosine”, although the phosphoryl group cannot be seen in the structure.)
We can get a better look at conformational differences, such as with the Abl A loop, by making an overlay of the two structures.
With the 2GQG structure already loaded, open the 1IEP structure in the same viewer window. Display both molecules as cartoons. In the command line type align 1IEP, 2GQG.
Locate residue 393 in both structures. It may help to view the structures in cartoon mode (like above) then display atoms only for the desired residues by selecting residue 393 in both molecules and to the right of (sele) click, S, sticks. You can also highlight the A loop of each structure in a different color.
20) How does the location of residue 393 differ in the two structures?
Find the inhibitor in each structure (for example by typing show sticks, organic) in the command line.
21) Very briefly compare the binding orientation of Gleevec and Dasatinib.
22) The H396P mutation in Abl destabilizes the inactive conformation of the kinase. Based on the Gleevec-bound and Dasatinib-bound structures of st Abl, explain how your laboratory results from the kinase activity assays with wt and H396P Abl are are consistent (or not consistant) with these structures. (If you have not yet completed Session 14, predict what you would expect to see in the H396P assays based structural evidence. Compare this to your actual results after completing your assays.)
Optional: If you are interested in exploring the structure of the H396P mutant, you can bring up the PDB file 2F4J, which is H396P Abl bound to another inhibitor, VX-680. This is NOT required to answer any of the questions above.
Note: To save a high quality image of any molecular structure from Pymol: 1) type bg_color white 2) type ray 1200,1200 3) select save image as and then select PNG.
SESSION 15 Answer Key
1) What type of secondary structure is most prominent in Abl? alpha helix
2) What type of secondary structure is most prominent in the N-lobe (residues 225-350) of Abl? beta sheet (ok to also include that there is an alpha helix)
3) What type of secondary structure is most prominent in the C-lobe (residues 354-498) of Abl? alpha helix
4) The protein contains a five-stranded β-sheet. Is this sheet located at the C or N terminus of the kinase domain? N-terminus
5) What residues comprise this beta-sheet? For this answer do not include the helix that is between the strands, so you should list two ranges. (For example “S229-G250 and L340-A350” would in the correct form- although this is not the correct answer). It may be helpful to zoom in on your structure to answer this question. I(242)-L(266) and L(301)-E(316)
6) Where are most of the hydrophobic residues located, on the inside or the outside of the protein? (You may want to switch back and forth from viewing the sticks to the cartoon form to get a better view of this.) on the inside
7) Where are most of the polar residues located? on the outside
8) Does this make sense in terms of protein folding? Explain. Yes. The residues on the outside of a protein will interact with water molecules, so it makes sense for them to be polar. The hydrophobic molecules are both shielded from the water and able to interact with each other on the inside of the protein.
9) When bound to Gleevec, is the A loop of the Abl kinase domain in the open (extended) or closed conformation? Very briefly explain your answer. Closed. The activation loop is blocking the substrate binding site and the Asp residue of the DFG motif is pointing away from the ATP binding site.
10) When bound to Gleevec, is Abl kinase domain in the active or inactive form. Very briefly explain your answer. Inactive. The A loop is closed in the inactive conformation.
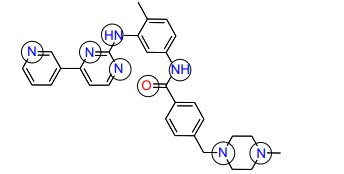
11) What groups on the structure of Gleevec above could participate in hydrogen bonds? (Draw Gleevec and indicate the groups by circling).
12) Looking at the Abl structure, name the residues that appear to hydrogen bond to Gleevec and give the distances between the atoms. Draw these on your Gleevec structure. Hint: there are 4 possible hydrogen bonds within 3.0 Å .
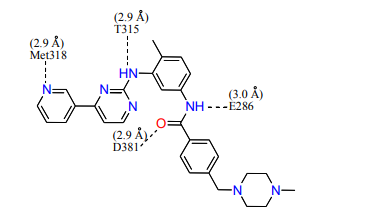
13) Name two other interactions that may be stabilizing the inhibitor in the active site. Van der Waals and hydrophobic interactions
For questions 14 through 18, consider the most common mutation site found in cases of Gleevec-resistant CML, site 315.
14) What amino acid is at this site, and what sort of interaction does this particular residue have with the inhibitor? Thr residue. H-bond.
15) What might you predict would happen if this residue were mutated to Asn or Ile? H-bond would not be possible and there would be steric interference with Gleevec binding.
16) What if this residue were mutated to Ala? H-bond not possible. No steric effect.
17) Is residue 315 interacting with Dasatinib? If so, describe the interaction. Yes. H-bond.
18) Would you expect Dasatinib to effectively inhibit an Abl mutant with residue 315 mutated to an Ile? Very briefly explain why or why not. No. H-bond would break and would sterically interefere with binding (although not as extremely as for Gleevec)
19) When bound to Dasatinib, is the A loop of the Abl kinase domain in the open (extended) or closed conformation? Very briefly explain your answer. Open. The activation loop away from the substrate binding site and the Asp residue of the DFG motif is pointing into the ATP binding site.
20) How does the location of residue 393 differ in the two structures? In the Gleevec-bound structure, Tyr393 is in the substrate binding pocket, and in the Dasatinib-bound structure, Tyr 393 is moved far away from the substrate binding pocket.
21) Very briefly compare the binding orientation of Gleevec and Dasatinib. Many answers are acceptable for this. For example, students could draw the parts of the two molecules that overlap and the parts that are pointing in opposite directions. Alternatively, they could write that Gleevec binds deep into a hydrophobic pocket, while Dasatinib binds more similarly to ATP (they would need to look at their Lecture 4 notes to see this).
22) The H396P mutation in Abl destabilizes the inactive conformation of the kinase. Based on the Gleevec-bound and Dasatinib-bound structures of wt Abl, explain how your laboratory results from the kinase activity assays with wt and H396P Abl are consistent (or not consistent) with these structures. (If you have not yet completed Session 14, predict what you would expect to see in the H396P assays based structural evidence. Compare this to your actual results after completing your assays.) The structures show that Gleevec binds the inactive form, and Dasatinib binds the active form. This is consistent with the kinase assays, which show that the wt Abl is inhibited by both Gleevec and Dasatinib (consistent with the fact that the wt can take on both the active and inactive forms), while H396P is only inhibited by Dasatinib (which makes sense because the inactive form is destabilized in this mutant).