
W2018_Bis2A_Lecture10_reading
( \newcommand{\kernel}{\mathrm{null}\,}\)
Overview
In this reading assignment, you are asked to reread and then comment on selected sections of the Lectures 2-9 reading assignments. The sections were chosen based on their high interest, level of confusion, number of questions, or the quality of the comments. Please note that this does NOT mean that these sections are the only ones that will appear on the test.
Many of these topics should be less confusing now that you have mastered the learning objectives from the first three weeks of the course. You should find it easier to make substantial comments on this material that reflect your deeper understanding of this material.
From the reading for Lecture 2
Diffusion and its importance to bacteria and archaea
Movement by diffusion is passive and proceeds down the concentration gradient. For compounds to move from the outside to the inside of the cell, the compound must be able to cross the phospholipid bilayer. If the concentration of a substance is lower inside the cell than outside and it has chemical properties that allow it to move across the cell membrane, that compound will energetically tend to move into the cell. While the "real" story is a bit more complex and will be discussed in more detail later, diffusion is one of the mechanisms bacteria and archaea use to aid in the transport of metabolites.
Diffusion can also be used to get rid of some waste materials. As waste products accumulate inside the cell, their concentration rises compared to that of the outside environment, and the waste product can leave the cell. Movement within the cell works the same way: compounds will move down their concentration gradient, away from where they are synthesized to places where their concentration is low and therefore may be needed. Diffusion is a random process—the ability of two different compounds or reactants for chemical reactions to interact becomes a meeting of chance. Therefore, in small, confined spaces, random interactions or collisions can occur more frequently than they can in large spaces.
The ability of a compound to diffuse depends on the viscosity of the solvent. For example, it is a lot easier for you to move around in air than in water (think about moving around underwater in a pool). Likewise, it is easier for you to swim in a pool of water than in a pool filled with peanut butter. If you put a drop of food coloring into a glass of water, it quickly diffuses until the entire glass has changed color. Now what do you think would happen if you put that same drop of food coloring into a glass of corn syrup (very viscous and sticky)? It will take a lot longer for the glass of corn syrup to change color.
The relevance of these examples is to note that the cytoplasm tends to be very viscous. It contains many proteins, metabolites, small molecules, etc. and has a viscosity more like corn syrup than water. So, diffusion in cells is slower and more limited than you might have originally expected. Therefore, if cells rely solely on diffusion to move compounds around, what do you think happens to the efficiency of these processes as cells increase in size and their internal volumes get bigger? Is there a potential problem to getting big that is related to the process of diffusion?
From the reading for Lecture 3
Water
Water is a unique substance whose special properties are intimately tied to the processes of life. Life originally evolved in a watery environment, and most of an organism’s cellular chemistry and metabolism occur inside the water-solvated contents of the cell. Water solvates or "wets" the cell and the molecules in it, plays a key role as reactant or product in an innumerable number of biochemical reactions, and mediates the interactions between molecules in and out of the cell. Many of water’s important properties derive from the molecule's polar nature, which can be tracked down to the polar molecules whose dipole originates from its polar covalent bonds between hydrogen and oxygen.
In BIS2A, the ubiquitous role of water in nearly all biological processes is easy to overlook by getting caught up in the details of specific processes, proteins, the roles of nucleic acids, and in your excitement for molecular machines (it'll happen). It turns out, however, that water plays key roles in all of those processes and we will need to continuously stay aware of the role that water is playing if we are to develop a more functional understanding. Be on the lookout and also pay attention when your instructor points this out.
In a liquid state, individual water molecule interact with one another through a network of dynamic hydrogen bonds that are being constantly forming and breaking. Water also interacts with other molecules that have charged functional groups and/or functional groups with hydrogen bond donors or acceptors. A substance with sufficient polar or charged character may dissolve or be highly miscible in water is referred to as being hydrophilic (hydro- = “water”; -philic = “loving”). By contrast, molecules with more nonpolar characters such as oils and fats do not interact well with water and separate from it rather than dissolve in it, as we see in salad dressings containing oil and vinegar (an acidic water solution). These nonpolar compounds are called hydrophobic (hydro- = “water”; -phobic = “fearing”). We will consider the some of the energetic components of these types of reactions in other another chapter.
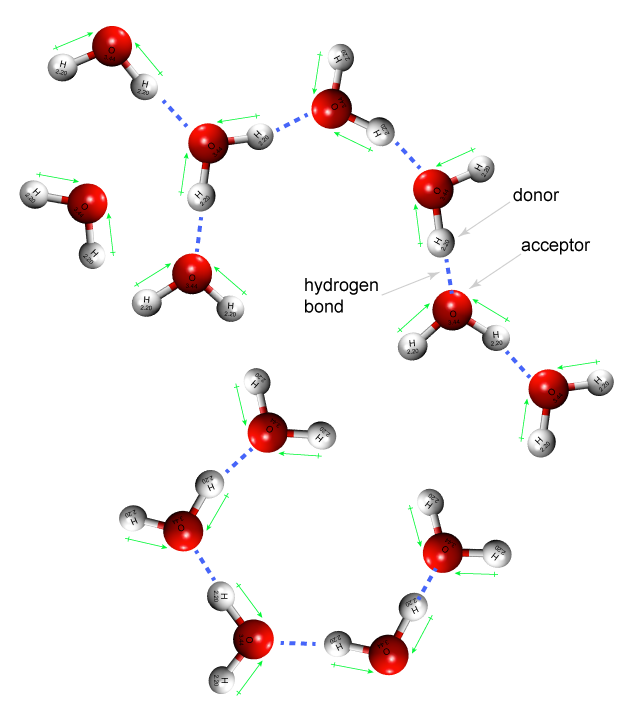
Figure 1. In a liquid state water forms a dynamic network of hydrogen bonds between individual molecules. Shown are one donor-acceptor pair.
Attribution: Marc T. Facciotti (original work)
Water's solvent properties
Since water is a polar molecule with slightly positive and slightly negative charges, ions and polar molecules can readily dissolve in it. Therefore, water is referred to as a solvent, a substance capable of dissolving other polar molecules and ionic compounds. The charges associated with these molecules will form hydrogen bonds with water, surrounding the particle with water molecules. This is referred to as a sphere of hydration, or a hydration shell and serves to keep the particles separated or dispersed in the water.
When ionic compounds are added to water, the individual ions interact with the polar regions of the water molecules, and the ionic bonds are likely disrupted in the process called dissociation. Dissociation occurs when atoms or groups of atoms break off from molecules and form ions. Consider table salt (NaCl, or sodium chloride). A dry block of NaCl is held together by ionic bonds and is difficult to dissociate. When NaCl crystals are added to water, however, the molecules of NaCl dissociate into Na+ and Cl– ions, and spheres of hydration form around the ions. The positively charged sodium ion is surrounded by the partially negative charge of the water molecule’s oxygen. The negatively charged chloride ion is surrounded by the partially positive charge of the hydrogen on the water molecule. One may imagine a model in which the ionic bonds in the crystal are "traded" for many smaller scale ionic bonds with the polar groups on water molecules.
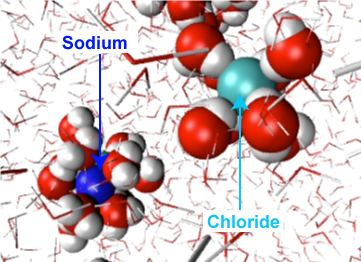
Figure 2. When table salt (NaCl) is mixed in water, spheres of hydration are formed around the ions. This figure depicts a sodium ion (dark blue sphere) and a chloride ion (light blue sphere) solvated in a "sea" of water. Note how the dipoles of the water molecules surrounding the ions are aligned such that complementary charges/partial charges are associating with one another (i.e., the partial positive charges on the water molecules align with the negative chloride ion whereas the partial negative charges on the oxygen of water align with the positively charged sodium ion).
Attribution: Ting Wang - UC Davis (original work modified by Marc T. Facciotti)
Note: possible discussion
Consider the model of water dissolving a salt crystal presented above. Describe in your own words how this model can be used to explain what is happening at the molecular level when enough salt is added to a volume of water that the salt no longer dissolves (the solution reaches saturation). Work together to craft a common picture.
From the reading for Lecture 4
pH
The pH of a solution is a measure of the concentration of hydrogen ions in a solution (or the number of hydronium ions). The number of hydrogen ions is a direct measure of how acidic or how basic a solution is. The concentration of hydrogen ions dissociating from pure water is 1 × 10-7 moles H+ ions per liter of water.
1 mole (mol) of a substance (which can be atoms, molecules, ions, etc), is defined as being equal to 6.02 x 1023 particles of the substance. Therefore, 1 mole of water is equal to 6.02 x 1023 water molecules. The pH is calculated as the negative of the base 10 logarithm of this unit of concentration. The log10 of 1 × 10-7 is -7.0, and the negative of this number yields a pH of 7.0, which is also known as neutral pH.
Non-neutral pH readings result from dissolving acids or bases in water. High concentrations of hydrogen ions yields a low pH number, whereas low levels of hydrogen ions result in a high pH.
This inverse relationship between pH and the concentration of protons confuses many students - take the time to convince yourself that you "get it."
An acid is a substance that increases the concentration of hydrogen ions (H+) in a solution, usually by having one of its hydrogen atoms dissociate. For example, we have learned that the carboxyl functional group is an acid. The hydrogen atom can dissociate from the oxygen atom resulting in a free proton and a negatively charged functional group. A base provides either hydroxide ions (OH–) or other negatively charged ions that combine with hydrogen ions, effectively reducing the H+ concentration in the solution and thereby raising the pH. In cases where the base releases hydroxide ions, these ions bind to free hydrogen ions, generating new water molecules. For example, we have learned that the amine functional group is a base. The nitrogen atom will accept hydrogen ions in solution, thereby reducing the number of hydrogen ions which raises the pH of the solution.

Note: possible discussion
Look at the functional groups in Figure 3. Identify the protonated and deprotonated form of each functional group. Is the protonated form of a functional always the form that carries a charge? Describe in your own words the relationship between pH and the amount of protonation found on a specific functional group? How might this related to the electronegativity of the molecules present in the functional group?
From the reading for Lecture 5
pKa
pKa is defined as the negative log10 of the dissociation constant of an acid, its Ka. Therefore, the pKa is a quantitative measure of how easily or how readily the acid gives up its proton [H+] in solution and thus a measure of the "strength" of the acid. Strong acids have a small pKa, weak acids have a larger pKa.
The most common acid we will talk about in BIS2A is the carboxylic acid functional group. These acids are typically weak acids, meaning that they only partially dissociate (into H+ cations and RCOO- anions) in neutral solution. HCL (hydrogen chloride) is a common strong acid, meaning that it will fully dissociate into H+ and Cl-.
Note that the key difference in the figure below between a strong acid or base and a weak acid or base is the single arrow (strong) versus a double arrow (weak). In the case of the single arrow you can interpret that by imagining that nearly all reactants have been converted into products. Moreover, it is difficult for the reaction to reverse backwards to a state where the protons are again associated with the molecule there were associated with before. In the case of a weak acid or base, the double-sided arrow can be interpreted by picturing a reaction in which:
- both forms of the conjugate acid or base (that is what we call the molecule that "holds" the proton - i.e. CH3OOH and CH3OO-, respectively in the figure) are present at the same time and
- the ratio of those two quantities can change easily by moving the reaction in either direction.

Figure 1. An example of strong acids and strong bases in their protonation and deprotonation states. The value of their pKa is shown on the left. Attribution: Marc T. Facciotti
Electronegativity plays a role in the strength of an acid. If we consider the hydroxyl group as an example, the greater electronegativity of the atom or atoms (indicated R) attached to the hydroxyl group in the acid R-O-H results in a weaker H-O bond, which is thus more readily ionized. This means that the pull on the electrons away from the hydrogen atom gets greater when the oxygen atom attached to the hydrogen atom is also attached to another electronegative atom. An example of this is HOCL. The electronegative Cl polarizes the H-O bond, weakening it and facilitating the ionization of the hydrogen. If we compare this to a weak acid where the oxygen is bound to a carbon atom (as in carboxylic acids) the oxygen is bound to the hydrogen and carbon atom. In this case, the oxygen is not bound to another electronegative atom. Thus the H-O bond is not further destabilized and the acid is considered a weak acid (it does not give up the proton as easily as a strong acid).

Figure 2. The strength of the acid can be determined by the electronegativity of the atom the oxygen is bound to. For example, the weak acid Acetic Acid, the oxygen is bound to carbon, an atom with low electronegativity. In the strong acid, Hypochlorous acid, the oxygen atom is bound to an even more electronegative Chloride atom.
Attribution: Erin Easlon
In Bis2A you are going to be asked to relate pH and pKa to each other when discussing the protonation state of an acid or base, for example, in amino acids. How can we use the information given in this module to answer the question: Will the functional groups on the amino acid Glutamate be protonated or deprotonated at a pH of 2, at a pH of 8, at a pH of 11?
In order to start answering this question we need to create a relationship between pH and pKa. The relationship between pKa and pH is mathematically represented by Henderson-Hasselbach equation shown below, where [A-] represents the deprotonated form of the acid and [HA] represents the protonated form of the acid.
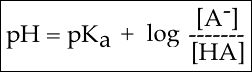
Figure 3. The Henderson-Hasselbach equation
A solution to this equation is obtained by setting pH = pKa. In this case, log([A-] / [HA]) = 0, and [A-] / [HA] = 1. This means that when the pH is equal to the pKa there are equal amounts of protonated and deprotonated forms of the acid. For example, if the pKa of the acid is 4.75, at a pH of 4.75 that acid will exist as 50% protonated and 50% deprotonated. This also means that as the pH rises, more of the acid will be converted into the deprotonated state and at some point the pH will be so high that the majority of the acid will exist in the deprotonated state.

Figure 4. This graph depicts the protonation state of acetic acid as the pH changes. At a pH below the pKa, the acid is protonated. At a pH above the pKa the acid is deprotonated. If the pH equals the pKa, the acid is 50% protonated and 50% deprotonated. Attribution: Ivy Jose
In BIS2A, we will be looking at the protonation state and deprotonation state of amino acids. Amino acids contain multiple functional groups that can be acids or bases. Therefore their protonation/deprotonation status can be more complicated. Below is the relationship between the pH and pKa of the amino acid Glutamic Acid. In this graph we can ask the question we posed earlier: Will the functional groups on the amino acid Glutamate be protonated or deprotonated at a pH of 2, at a pH of 8, at a pH of 11?
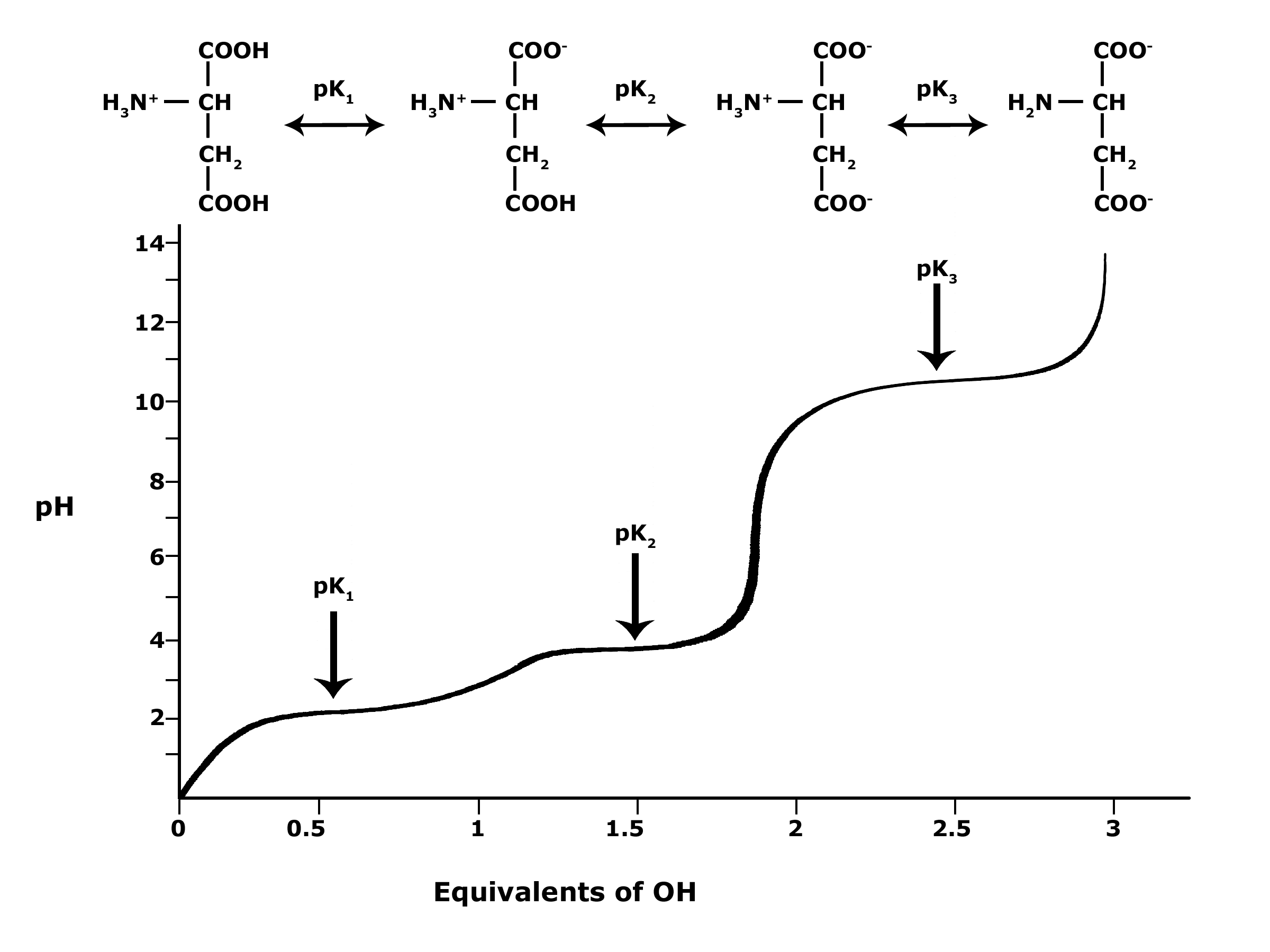
Figure 5. This graph depicts the protonation state of glutamate as the pH changes. At a pH below the pKa for each functional group on the amino acid, the functional group is protonated. At a pH above the pKa for the functional group it is deprotonated. If the pH equals the pKa, the functional group is 50% protonated and 50% deprotonated.
Attribution: Ivy Jose
Note: Possible discussion
- What is the overall charge of free Glutamate at a pH of 5?
- What is the overall charge of free Glutamate at a pH of 10?
From the reading for Lecture 6
Reversibility
In theory, any chemical reaction can proceed in either direction under the right conditions. Reactants may synthesize into a product that later reverts back to a reactant. Reversibility is also a quality of exchange reactions. For instance, A+BC→AB+C could then reverse to AB+C→A+BC. This reversibility of a chemical reaction is indicated with a double arrow: A+BC⇄AB+C.
Synthesis reactions
Many macromolecules are made from smaller subunits, or building blocks, called monomers. Monomers covalently link to form larger molecules known as polymers. Often, the synthesis of polymers from monomers will also produce water molecules as products of the reaction. This type of reaction is known as dehydration synthesis or condensation reaction.
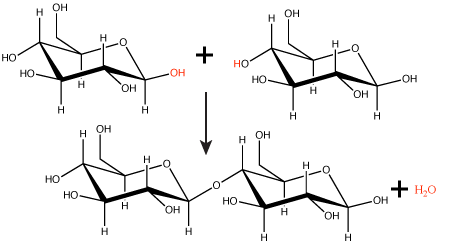
Figure 1. In the dehydration synthesis reaction depicted above, two molecules of glucose are linked together to form the disaccharide maltose. In the process, a water molecule is formed.
Attribution: Marc T. Facciotti (original work)
In a dehydration synthesis reaction (Figure 1), the hydrogen of one monomer combines with the hydroxyl group of another monomer, releasing a molecule of water. At the same time, the monomers share electrons and form covalent bonds. As additional monomers join, this chain of repeating monomers forms a polymer. Different types of monomers can combine in many configurations, giving rise to a diverse group of macromolecules. Even one kind of monomer can combine in a variety of ways to form several different polymers; for example, glucose monomers are the constituents of starch, glycogen, and cellulose.
In the carbohydrate monomer example above, the polymer is formed by a dehydration reaction; this type of reaction is also used to add amino acids to a growing peptide chain and nucleotides to the growing DNA or RNA polymer. Visit the modules on Amino Acids, Lipids, and Nucleic Acids to see if you can identify the water molecules that are removed when a monomer is added to the growing polymer.
![]()
Figure 2. This depicts, using words, (decorated with functional groups colored in red) a generic dehydration synthesis/condensation reaction.
Attribution: Marc T. Facciotti (original work)
Hydrolysis reactions
Polymers are broken down into monomers in a reaction known as hydrolysis. A hydrolysis reaction includes a water molecule as a reactant (Figure 3). During these reactions, a polymer can be broken into two components: one product carries a hydrogen ion (H+) from the water, while the second product carries the water's remaining hydroxide (OH–).
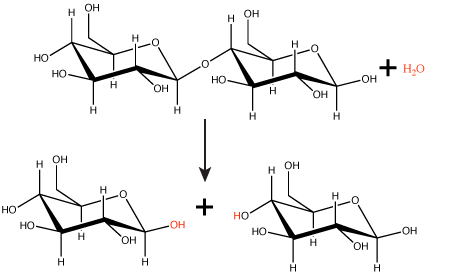
Figure 3. In the hydrolysis reaction shown here, the disaccharide maltose is broken down to form two glucose monomers with the addition of a water molecule. Note that this reaction is the reverse of the synthesis reaction shown in Figure 1 above.
Attribution: Marc T. Facciotti (original work)
![]()
Figure 4. This depicts using words (decorated with functional groups colored in red) a generic hydrolysis reaction.
Attribution: Marc T. Facciotti (original work)
Dehydration synthesis and hydrolysis reactions are catalyzed, or “sped up,” by specific enzymes. Note that both dehydration synthesis and hydrolysis reactions involve the making and breaking of bonds between the reactants—a reorganization of the bonds between the atoms in the reactants. In biological systems (our bodies included), food in the form of molecular polymers is hydrolyzed into smaller molecules by water via enzyme-catalyzed reactions in the digestive system. This allows for the smaller nutrients to be absorbed and reused for a variety of purposes. In the cell, monomers derived from food may then be reassembled into larger polymers that serve new functions.
Helpful links: Visit this site to see visual representations of dehydration synthesis and hydrolysis.
From the reading for Lecture 7
Free Energy
If we want to describe transformations, it is useful to have a measure of (a) how much energy is in a system, (b) the dispersal of that energy within the system and, of course, (c) how these change between the start and end of a process. The concept of free energy, often referred to as Gibbs free energy or free enthalpy (abbreviated with the letter G), in some sense, does just that. Gibbs free energy can be defined in several interconvertible ways, but a useful one in the context of biology is the enthalpy (internal energy) of a system minus the entropy of the system scaled by the temperature. The difference in free energy when a process takes place is often reported in terms of the change (Δ) of enthalpy (internal energy) denoted H, minus the temperature scaled change (Δ) in entropy, denoted S. See the equation below.
ΔG=ΔH−TΔS
The Gibbs energy is often interpreted as the amount of energy available to do useful work. With a bit of hand waving, we can interpret this by invoking the idea presented in the section on entropy, which states the dispersion of energy (required by the Second Law) associated with a positive change in entropy somehow renders some of the energy that is transferred less useful to do work. One can say that this is reflected in part in the T∆S term of the Gibbs equation.
To provide a basis for fair comparisons of changes in Gibbs free energy amongst different biological transformations or reactions, the free energy change of a reaction is measured under a set of common standard experimental conditions. The resulting standard free energy change of a chemical reaction is expressed as an amount of energy per mole of the reaction product (either in kilojoules or kilocalories, kJ/mol or kcal/mol; 1 kJ = 0.239 kcal), when measured at a standard pH, temperature, and pressure conditions. Standard pH, temperature, and pressure conditions are generally standardized at pH 7.0, 25 degrees Celsius, and 100 kilopascals (1 atm pressure), respectively. It is important to note that cellular conditions vary considerably from these standard conditions, and so actual ∆G inside a cell will differ considerably from those calculated under standard conditions.
Endergonic and exergonic reactions
For reactions with ∆G < 0, the products of the reaction have less free energy than the reactants. Since ∆G is the difference between the enthalpy and entropy changes in a reaction, a net negative ∆G can arise in different ways. The left panel of Figure 1 below shows a common graphical representation of an exergonic reaction. Free energy is plotted on the y-axis, and the x-axis in arbitrary units shows the progress of a reaction. This type of graph is called a reaction coordinate diagram. In the case of an exergonic reaction, the figure indicates two key things: (1) the difference between the free energy of the reactants and products is negative and (2) the progress of the reaction requires some input of free energy (shown as an energy hill). This graph does not tell us how the energy in the system was redistributed, only that the difference between enthalpy and entropy is negative. Reactions that have a negative ∆G are termed exergonic reactions. These reactions are said to occur spontaneously. Understanding which chemical reactions are spontaneous is extremely useful for biologists who are trying to understand whether a reaction is likely to "go" or not.
It is important to note that the term "spontaneous"—in the context of thermodynamics—does NOT imply anything about how fast the reaction proceeds. The change in free energy only describes the difference between beginning and end states, NOT how fast that transition takes place. This is somewhat contrary to the everyday use of the term, which usually carries the implicit understanding that something happens quickly. As an example, the oxidation/rusting of iron is a spontaneous reaction. However, an iron nail exposed to air does not rust instantly—it may take years.
A chemical reaction with a positive ∆G means that the products of the reaction have a higher free energy than the reactants (see the right panel of Figure 1). These chemical reactions are called endergonic reactions, and they are NOT spontaneous. An endergonic reaction will not take place on its own without the transfer of energy into the reaction or increase of entropy somewhere else.
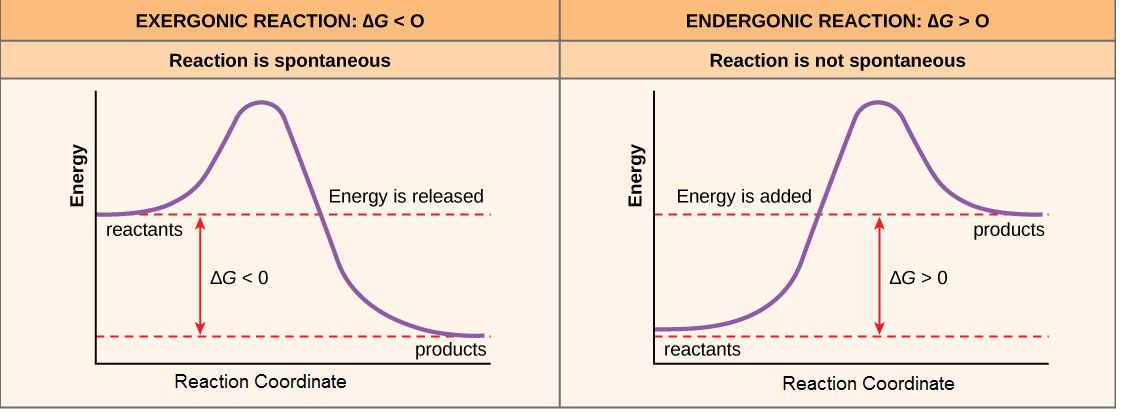
Figure 1. Exergonic and endergonic reactions result in changes in Gibbs free energy. In an exergonic reaction, the free energy of the products is lower than that of the reactants; meanwhile, in an endergonic reaction, the free energy of the products is higher than that of the reactants. Attribution: Marc T. Facciotti (own work)
The building of complex molecules, such as sugars, from simpler ones is an anabolic process and is endergonic. On the other hand, the catabolic process, such as the breaking down of sugar into simpler molecules, is generally exergonic. Like the example of rust above, while the breakdown of biomolecules is generally spontaneous, these reactions don’t necessarily occur instantaneously (quickly). Remember, the terms endergonic and exergonic only refer to the difference in free energy between the products and reactants; they don't tell you about the rate of the reaction (how fast it happens). The issue of rate will be discussed in later sections.
An important concept in the study of metabolism and energy is that of chemical equilibrium. Most chemical reactions are reversible. They can proceed in both directions, often transferring energy into their environment in one direction and transferring energy in from the environment in the other direction. The same is true for the chemical reactions involved in cell metabolism, such as the breaking down and building up of proteins into and from individual amino acids, respectively. Reactants within a closed system will undergo chemical reactions in both directions until a state of equilibrium is reached. This state of equilibrium is one of the lowest possible free energy states and is a state of maximal entropy. Equilibrium in a chemical reaction is the state in which both reactants and products are present in concentrations that have no further tendency to change with time. Usually, this state results when the forward reaction proceeds at the same rate as the reverse reaction. NOTE THIS LAST STATEMENT! Equilibrium means that the relative concentrations of reactants and products are not changing in time, BUT it does NOT mean that there is no interconversion between substrates and products—it just means that when the reactant(s) are converted to product(s) that product(s) are converted to reactant(s) at an equal rate (see Figure 2).
Either a rebalancing of substrate or product concentrations (by adding or removing substrate or product) or a positive change in free energy, typically by the transfer of energy from outside the reaction, is required to move a reaction out of a state of equilibrium. In a living cell, most chemical reactions do not reach a state of equilibrium—this would require that they reach their lowest free energy state. Energy is therefore required to keep biological reactions out of their equilibrium state. In this way, living organisms are in a constant, energy-requiring, uphill battle against equilibrium and entropy.

Figure 2. At equilibrium, do not think of a static, unchanging system. Instead, picture molecules moving in equal amounts from one area to another. Here, at equilibrium, molecules are still moving from left to right and right to left. The net movement however, is equal. There will still be about 15 molecules in each side of this flask once equilibrium is reached. Source: https://courses.candelalearning.com/...apter/entropy/
From the reading for Lecture 8
Enzymes
A substance that helps a chemical reaction to occur is a catalyst, and the special molecules that catalyze biochemical reactions are called enzymes. Almost all enzymes are proteins, made up of chains of amino acids, and they perform the critical task of lowering the activation energies of chemical reactions inside the cell. Enzymes do this by binding to the reactant molecules, and holding them in such a way as to make the chemical bond-breaking and bond-forming processes take place more readily. It is important to remember that enzymes don’t change the ∆G of a reaction. In other words, they don’t change whether a reaction is exergonic (spontaneous) or endergonic. This is because they don’t change the free energy of the reactants or products. They only reduce the activation energy required to reach the transition state.
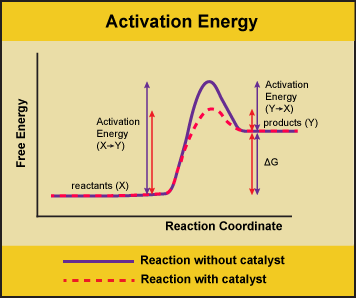
Figure 1: Enzymes lower the activation energy of the reaction but do not change the free energy of the reaction. Here the solid line in the graph shows the energy required for reactants to turn into products without a catalyst. The dotted line shows the energy required using a catalyst. This figure should say Gibbs Free Energy on the Y-axis and instead of noting deltaH should have deltaG. Attribution: Marc T. Facciotti (own work)
Enzyme Active Site and Substrate Specificity
The chemical reactants to which an enzyme binds are the enzyme’s substrates. There may be one or more substrates, depending on the particular chemical reaction. In some reactions, a single-reactant substrate is broken down into multiple products. In others, two substrates may come together to create one larger molecule. Two reactants might also enter a reaction, both become modified, and leave the reaction as two products. The location within the enzyme where the substrate binds is called the enzyme’s active site. The active site is where the “action” happens, so to speak. Since enzymes are proteins, there is a unique combination of amino acid residues (also called side chains, or R groups) within the active site. Each amino acid side-chain is characterized by different properties. Amino acids can be classified as large or small, weakly acidic or basic, hydrophilic or hydrophobic, positively or negatively charged, or neutral. The unique combination of amino acids, their positions, sequences, structures, and properties, creates a very specific chemical environment within the active site. This specific environment is suited to bind, albeit briefly, to a specific chemical substrate (or substrates). Due to this jigsaw puzzle-like match between an enzyme and its substrates (which adapts to find the best fit between the transition state and the active site), enzymes are known for their specificity. The “best fit” between an enzyme and its substrates results from the their respective shapes and the chemical complementarity of the functional groups on each binding partner.
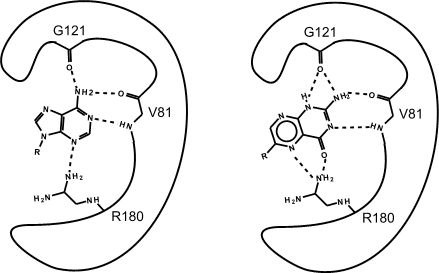
Figure 2: This is an enzyme with two different substrates bound in the active site. The enzymes are represented as blobs, except for the active site which shows the three R-groups of each of the three amino acids located in the active site. These R groups are interacting with the substrates through hydrogen bonding (represented as dashed lines)
At this point in the class you should be familiar with all the types of bonds as well as the chemical characteristics of all the functional groups. For example, the R group of R180 in the enzyme depicted above is the amino acid Arginine (abbreviated as R) and has an R group that consists of several amino functional groups. Amino functional groups contain a nitrogen (N) and hydrogen (H) atoms. Nitrogen is more electronegative than hydrogen so the covalent bond between N-H is a polar covalent bond. The hydrogen atoms in this bond will have a positive dipole moment, and the nitrogen atom will have a negative dipole moment. This allows amino groups to form hydrogen bonds with other polar compounds. Likewise, the backbone carbonyl oxygens of Valine (V) 81 and Glycine (G) 121 the backbone amino hydrogen of V81 are depicted engaged in hydrogen bonds with the small molecule substrate.
Prepare for the Test
Look to see which atoms in the figure above are involved in the hydrogen bonds between the amino acid R groups and the substrate. You will need to be able to identify these on your own, hydrogen bonds may not be drawn in for you on the test.
If you changed the pH of the solution that this enzyme was located in, would the enzyme still be able to form hydrogen bonds with the substrate ?
Which substrate (the left or right one) do you think is more stable in the active site? Why? How?
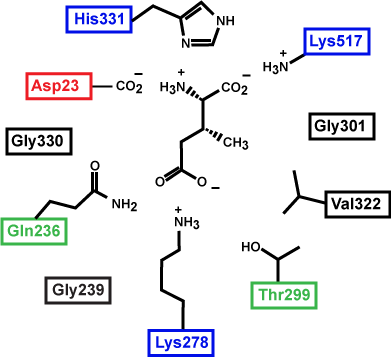
Figure 3: This is an enzyme active site. Only the amino acids in the active site are drawn. The substrate is sitting directly in the center. Source: Created by Marc T. Facciotti (original work)
Note
Prepare for the test: First, identify the type of macromolecule in the figure above. Second, draw in and label the appropriate interactions between the R groups and the substrate. Explain how these interactions might change if the pH of the solution changed.
Induced Fit and Enzyme Function
For many years, scientists thought that enzyme-substrate binding took place in a simple “lock-and-key” fashion. This model asserted that the enzyme and substrate fit together perfectly in one instantaneous step. However, current research supports a more refined view called induced fit. The induced-fit model expands upon the lock-and-key model by describing a more dynamic interaction between enzyme and substrate. As the enzyme and substrate come together, their interaction causes a mild shift in the enzyme’s structure that confirms an more productive binding arrangement between the enzyme and the transition state of the substrate. This energetically favorable binding maximizes the enzyme’s ability to catalyze its reaction.
When an enzyme binds its substrate, an enzyme-substrate complex is formed. This complex lowers the activation energy of the reaction and promotes its rapid progression in one of many ways. On a basic level, enzymes promote chemical reactions that involve more than one substrate by bringing the substrates together in an optimal orientation. The appropriate region (atoms and bonds) of one molecule is juxtaposed to the appropriate region of the other molecule with which it must react. Another way in which enzymes promote the reaction of their substrates is by creating an energetically favorable environment within the active site for the reaction to occur. Certain chemical reactions might proceed best in a slightly acidic or non-polar environment. The chemical properties that emerge from the particular arrangement of amino acid residues within an active site create the energetically favorable environment for an enzyme’s specific substrates to react.
The activation energy required for many reactions includes the energy involved in slightly contorting chemical bonds so that they can more easily react. Enzymatic action can aid this process. The enzyme-substrate complex can lower the activation energy by contorting substrate molecules in such a way as to facilitate bond-breaking. Finally, enzymes can also lower activation energies by taking part in the chemical reaction itself. The amino acid residues can provide certain ions or chemical groups that actually form covalent bonds with substrate molecules as a necessary step of the reaction process. In these cases, it is important to remember that the enzyme will always return to its original state at the completion of the reaction. One of the hallmark properties of enzymes is that they remain ultimately unchanged by the reactions they catalyze. After an enzyme is done catalyzing a reaction, it releases its product(s).
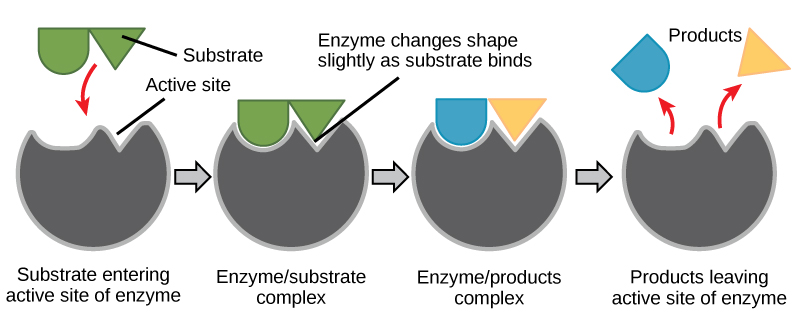
Figure 6: According to the induced-fit model, both enzyme and substrate undergo dynamic conformational changes upon binding. The enzyme contorts the substrate into its transition state, thereby increasing the rate of the reaction.
Creating an Energy story for the reaction above
Using the figure above, answer the questions posed in the energy story.
1. What are the reactants? What are the products?
2. What work was accomplished by the enzyme?
3. What state is the energy in initially? What state is the energy transformed into in the final state? This one might be tricky still, but try to identify where the energy is in the initial state and the final state.
Enzyme Regulation
Why regulate enzymes?
Cellular needs and conditions vary from cell to cell, and change within individual cells over time. The required enzymes and energetic demands of stomach cells are different from those of fat storage cells, skin cells, blood cells, and nerve cells. Furthermore, a digestive cell works much harder to process and break down nutrients during the time that closely follows a meal compared with many hours after a meal. As these cellular demands and conditions vary, so do the needed amounts and functionality of different enzymes.
Regulation of Enzymes by Molecules
Enzymes can be regulated in ways that either promote or reduce their activity. There are many different kinds of molecules that inhibit or promote enzyme function, and various mechanisms exist for doing so. In some cases of enzyme inhibition, for example, an inhibitor molecule is similar enough to a substrate that it can bind to the active site and simply block the substrate from binding. When this happens, the enzyme is inhibited through competitive inhibition, because an inhibitor molecule competes with the substrate for active site binding. On the other hand, in noncompetitive inhibition, an inhibitor molecule binds to the enzyme in a location other than an allosteric site and still manages to block substrate binding to the active site.
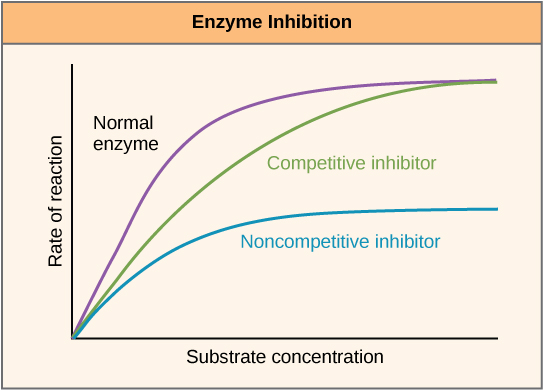
Figure 7: Competitive and noncompetitive inhibition affect the rate of reaction differently. Competitive inhibitors affect the initial rate but do not affect the maximal rate, whereas noncompetitive inhibitors affect the maximal rate.
Some inhibitor molecules bind to enzymes in a location where their binding induces a conformational change that reduces the affinity of the enzyme for its substrate. This type of inhibition is called allosteric inhibition. Most allosterically regulated enzymes are made up of more than one polypeptide, meaning that they have more than one protein subunit. When an allosteric inhibitor binds to an enzyme, all active sites on the protein subunits are changed slightly such that they bind their substrates with less efficiency. There are allosteric activators as well as inhibitors. Allosteric activators bind to locations on an enzyme away from the active site, inducing a conformational change that increases the affinity of the enzyme’s active site(s) for its substrate(s).
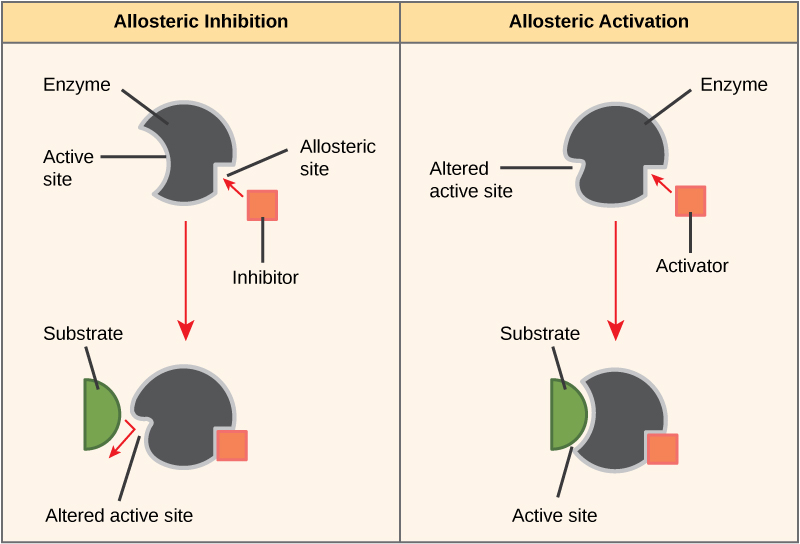
Figure 8: Allosteric inhibitors modify the active site of the enzyme so that substrate binding is reduced or prevented. In contrast, allosteric activators modify the active site of the enzyme so that the affinity for the substrate increases.
Video Link
Check out this short (1 minute) video on competitive vs. noncompetitive enzymatic inhibition. Also, take a look at this video (1.2 minutes) on feed back inhibition.
Many enzymes don’t work optimally, or even at all, unless bound to other specific non-protein helper molecules, either temporarily through ionic or hydrogen bonds or permanently through stronger covalent bonds. Two types of helper molecules are cofactors and coenzymes. Binding to these molecules promotes optimal conformation and function for their respective enzymes. Cofactors are inorganic ions such as iron (Fe2+) and magnesium (Mg2+). One example of an enzyme that requires a metal ion as a cofactor is the enzyme that builds DNA molecules, DNA polymerase, which requires bound zinc ion (Zn2+) to function. Coenzymes are organic helper molecules, with a basic atomic structure made up of carbon and hydrogen, which are required for enzyme action. The most common sources of coenzymes are dietary vitamins. Some vitamins are precursors to coenzymes and others act directly as coenzymes. Vitamin C is a coenzyme for multiple enzymes that take part in building the important connective tissue component, collagen. An important step in the breakdown of glucose to yield energy is catalysis by a multi-enzyme complex called pyruvate dehydrogenase. Pyruvate dehydrogenase is a complex of several enzymes that actually requires one cofactor (a magnesium ion) and five different organic coenzymes to catalyze its specific chemical reaction. Therefore, enzyme function is, in part, regulated by an abundance of various cofactors and coenzymes, which are supplied primarily by the diets of most organisms.
Enzyme Compartmentalization
In eukaryotic cells, molecules such as enzymes are usually compartmentalized into different organelles. This allows for yet another level of regulation of enzyme activity. Enzymes required only for certain cellular processes can be housed separately along with their substrates, allowing for more efficient chemical reactions. Examples of this sort of enzyme regulation based on location and proximity include the enzymes involved in the latter stages of cellular respiration, which take place exclusively in the mitochondria, and the enzymes involved in the digestion of cellular debris and foreign materials, located within lysosomes.
Additional Links
The following links will take you to a series of videos on kinetics. The first link contains 4 videos on reaction rates and the second link contains 9 videos related to the relationship between reaction rates and concentration. These videos are supplemental and are provided to give you an outside resource to further explore enzyme kinetics.
Tags recommended by the template: article:topic