12.7: Actin - Myosin Structures in Muscle
- Page ID
- 19240
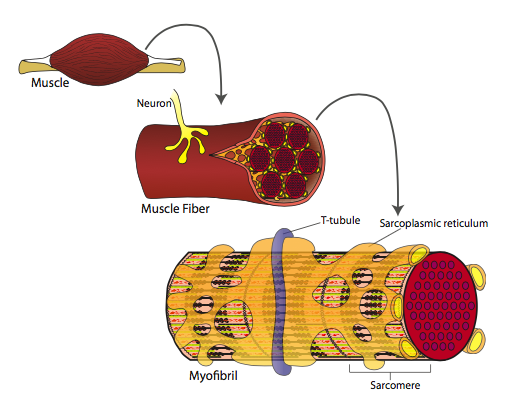
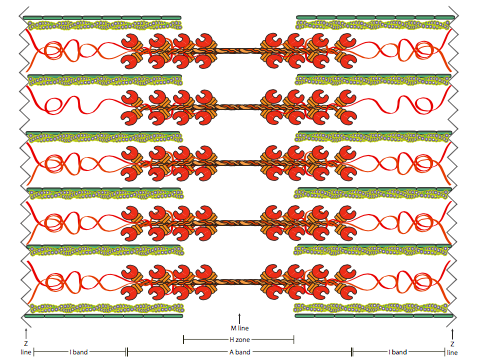
The motor proteins that transport materials along the acting microfilaments are similar in some ways, such as the globular head group that binds and hydrolyzes ATP, yet different in other ways, such as the motion catalyzed by the ATP hydrolysis. Much of the f-actin and myosin in striated and cardiac muscle cells is found in a peculiar arrangement designed to provide a robust contractile response over the entire length of the cell. The sarcomere is an arrangement of alternating fibers of f-actin (also known as “thin fibers” based on their appearance in electron micrographs) and myosin II (or “thick fibers”). Although we do not normally think of the motor protein as a fiber, in this case the tails of the myosin II molecules intertwine to form a continuous fiber of myosin molecules. As the contractile cycle proceeds, the myosin molecules grip the adjacent actin fibers, and move them. In Figure \(\PageIndex{11}\), you can see that a sarcomere is constructed so that the stationary myosin fibers are located centrally, with two parallel sets of actin fibers interspersed between the myosin fibers, to the left and the right of the center. Note that the actin fibers do not cross the center line, and that at the center, the myosin molecules switch orientation. The physiological effect of this is that the actin filaments are all pulled inwards toward the center of the sarcomere. The sarcomere in turn, is merely one of many connected together to form a myofibril. The myofibrils extend the length of the muscle cell.

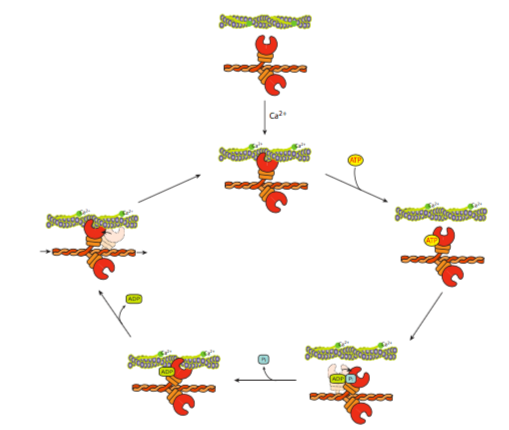
When the myosin head is in its resting state, it is tightly attached to the actin filament. In fact, rigor mortis occurs in dead animals because there is no more ATP being made, and thus the sarcomeres are locked into place. Rigor begins approximately 2-3 hours after death in humans, after reserves of ATP are depleted. When the body relaxes again in about 3 days, it is due to the decomposition and breakdown of the actin and myosin proteins. However, while they are still living animals, ATP is generally available, and it can bind to the myosin head, causing it to lose affinity for the f-actin, and let go (Figure \(\PageIndex{12}\)). At this point, no significant movement has occurred. Once the ATP is hydrolyzed though, the myosin head can reattach to the f-actin a little further down the filament than it had originally. The energy released is stored in the neck region. The ADP and Pi are still attached to the myosin head as well. The next step is for the Pi to drop off the myosin, leading to the power stroke. The neck of the myosin swivels around, leading to a translocation of the head by approximately 10 nm for myosin II. The distance of translocation varies depending on the type of myosin, but it is not yet clear whether the length of the neck is proportional to the displacement of the head. Finally, the ADP drops off the myosin head, increasing the affinity of the head for the f-actin.
The sarcomere structure described in the first paragraph was incomplete in order to place the major players clearly in their roles. There are other proteins in the sarcomere with important structural and regulatory functions. One of the key regulatory components is tropomyosin. This is a fibrous protein that lies in the groove of an actin microfilament and blocks access to the myosin binding site. Tropomyosin attaches to the microfilament in conjunction with a multi-subunit troponin complex. When Ca2+ is available, it can bind to troponin-C, leading to a conformational change that shifts the position of tropomyosin to reveal the myosin binding site. This is the primary point of control for muscle contraction: recall that intracellular Ca2+ levels are kept extremely low because its primary function is in intracellular signaling. One way that the Ca2+ levels are kept that low is to pump it into a reservoir, such as the endoplasmic reticulum.
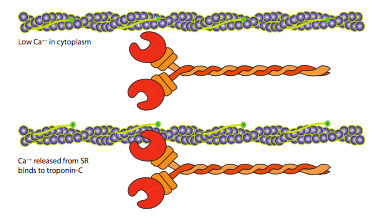
In muscle cells, there is a specialization of the ER called the sarcoplasmic reticulum (SR) that is rich in Ca2+ pumps and Ca2+. When a signal is sent from a controlling nerve cell to the muscle cell, it causes a depolarization of the muscle cell membrane. This consequently depolarizes a set of membranes called the transverse tubules (T-tubules) that lie directly on parts of the sarcoplasmic reticulum. There are proteins on the t-tubule surface that directly interact with a set of Ca2+ channel proteins, holding the channel closed normally. When the t-tubule is depolarized, the proteins change shape, which changes the interaction with the Ca2+ channels on the SR, and allows them to open. Ca2+ rushes out of the SR where it is available to troponin-c. Troponin-C bound to Ca2+ shifts the tropomyosin away from the actin filament, and the myosin head can bind to it. ATP can bind the myosin head to start the power stroke cycle, and voila, we have controlled muscle cell contraction.
The SR is a specialization of part of the endoplasmic reticulum, and contains a high concentration of Ca2+ ions because the SR membrane is embedded with Ca2+ pumps (ATPases) to keep the cytoplasmic concentration low and sequester the Ca2+ ions inside the SR. This is regulated by phosphorylation and [Ca2+] via a regulatory protein such as phospholamban (in cardiac muscle). Phospholamban is an integral membrane protein of the SR that normally associates with and inhibits the Ca2+ pump. However when it is phosphorylated, or as cytoplasmic Ca2+ levels rise, the phospholamban releases from the Ca2+ pump and allows it to function.
In addition to the “moving parts”, there are also more static, structural, proteins in the sarcomere (Figure \(\PageIndex{11}\)). Titin is a gigantic protein (the largest known, at nearly 3 MDa), and can be thought of as something of a bungee cord tether to the myosin fiber. Its essential purpose is to prevent the forces generated by the myosin from pulling the fiber apart. Titin wraps around the myosin fiber and attaches at multiple points, with the most medial just near the edge of the H zone. At the Z-line, titin attaches to a telethonin complex, which attach to the Z-disk proteins (antiparallel α-actinin). Titin also interacts with obscurin in the I-band region, where it may link myofibrils to the SR, and in the M-band region it can interact with the Ca2+-binding protein calmodulin-1 and TRIM63, thought to acts as a link between titin and the microtubule cytoskeleton. There are multiple isoforms of titin from alternative splicing, with most of the variation coming in the I-band region.
Disturbances to the proper formation of the titin-based support structure can be a cause of dilated cardiomyopathy, and from that, congestive heart failure. Some 20-30% of cases of dilated cardiomyopathy are familial, and mutations have been mapped to the N-terminal region of titin, where the protein interacts with telethonin. Defects in titin are also being investigated with respect to chronic obstructive pulmonary disease, and some types of muscular dystrophy.
Of course in an actual muscle (Figure \(\PageIndex{14}\)), what happens is that nerves grow into the muscle and make synaptic connections with them. At these synaptic connections, the nerve cell releases neurotransmitters such as acetylcholine (ACh), which bind to receptors (AChR) on the muscle cell. This then opens ion channels in the muscle cell membrane, triggering a voltage change across that membrane, which also happens to affect the nearby membrane of the transverse tubules subsequently opening Ca2+ channels in the SR. The contraction of sarcomeres can then proceed as already described above.