
D11. Chromatin Remodeling and Gene Expression
- Page ID
- 4995

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
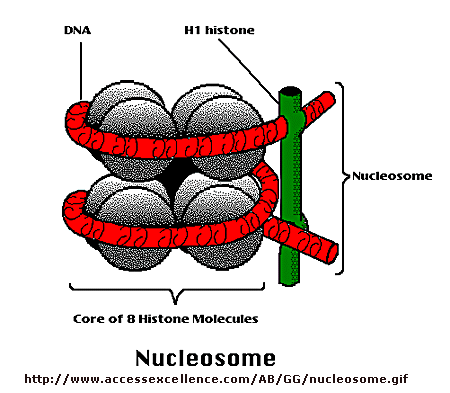
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Control of DNA transcription in eukaryotes was thought to involve the assembly of many proteins at the promoter into a pre-initiation complex (PIC). Once assembled, RNA polymerase could bind and transcription would be initiated. But wait a minute! Isn't DNA packaged in the nucleus into chromatin in which 147 BP of DNA is wound around a core of 4 pairs of positively charged histone proteins - including H2A, 2B, 3, and 4 - to form a nucleosome, seen under a microscope as beads on a string?
{kind=link}
{kind=link}
 Jmol: Updated Nucleosome Jmol14 (Java) | JSMol (HTML5)
Jmol: Updated Nucleosome Jmol14 (Java) | JSMol (HTML5)
Isn't this chromatin further wound into fibers which result in the classic picture of sister chromatids ready to separate at cell division?How could the transcription factors and RNA polymerase recognize target sites on DNA given this degree of "folding" and condensation of the DNA?
 Animation: Packing of DNA from the Walter + Eliza Hall Institute of Medical Research
Animation: Packing of DNA from the Walter + Eliza Hall Institute of Medical Research
Clearly the complex compacted state of DNA and its interaction with the histone proteins must be "remodeled" to allow interactions of the transcription factors and RNA polymerase (which is about the same size as a nucleosome). The regulation of this chromatin remodeling clearly affects gene transcription, and is another example of epigenetic changes that can affects phenotype. The state of chromatin structure is regulated by enzymes that affect histone structure and function by chemically modifying the histone proteins (through acetylation, methylation, and phosphorylation) . Likewise, the DNA at the promoter region is changed by enzymes that remodel the DNA through an ATP dependent series of modifications. For example when histones are modified by histone acetyltransferase (HAT's), other modeling factors (SWI/SNF) are recruited to the chromatin. Chromatin remodeling would also be affected by that cell cycle stage of the cell. For example, chromatin condensed in sister chromatids ready for cells division would have different remodeling requirements for gene transcription than might chromatin in the form of bead on a string. Likewise remodeling efforts would also be gene-specific.
The figure below shows how remodeling is coupled to formation of the pre-initiation complex for three genes:
- yeast HO gene: Swi5p activator binding results in the interaction of the SWI/SNF ATP-dependent remodeling enzyme, which leads to the binding of histone acetyltransferase (HAT). These facilitate formation of the pre-initiation complex.
- human interferon-β gene: gene sequences known as activators, 5' to the promoter, bind HATs. When histones are acetylated, SWI/SNF interacts to remodel the chromatin and facilitate PIC formation.
- human α-1 antitrypsin gene: the PIC is preformed and recruits HAT and SWI/SNF, which leads to gene transcription.
Alternations in chromatin remodeling could lead to changes in gene expression, in some cases causing cancer. SNF5 is a component of the SWI/SNF complex and in its normal form acts to suppress tumors (i.e. its gene is a tumor suppressor gene). Mutations in SNF5 are associated with rare and aggressive childhood tumors. Stuart Orkin has developed a technique to alter the gene in some mouse cells to produce an inverted gene which produces no functional SNF5. Cells with this mutation become tumor cells almost immediately.
Figure: Remodeling of Chromatin and Control of DNA Transcription

- SNF5 and cancer
DNA winds around the histone core to form the nucleosome. However, histone tails not associated with DNA binding protrude from the nucleosome, and the function of these tails is just being unraveled. The amino acids in these tails are clearly sites for posttranslational modifications, including methylation, acetylation, and phosphorylation. When modified, these tails would provide additional binding sites for protein which could regulate transcription and chromatin modeling, thus modifying the "genetic code". Understanding the "histone code" and how it affects gene transcription becomes important. For example, the methylation of Lys 9 on histone 3 leads to binding of heterochromatin-associated protein, leading to inhibition of gene transcription (an example of epigenetic silencing). Acetylation of the tails generally leads to activation of gene transcription at that site. Acetylation of Lys residues converts them to amides and removes the positive charge of the amine. This would lead to decreased electrostatic interactions between the DNA and histones proteins, making the DNA more available for interaction with transcription factors and RNA polymerase.
Epigenetic changes (through methylation of DNA or acetylation, methylation, and phosphorylation of histone proteins) causing chromatin remodeling may change phenotype (characteristics of the individual) as evidenced by the fact that identical twins can eventually diverge in ways that effect their propensities to disease. Differences in diet and lifestyle, which can alter disease propensity, might exert their effects through epigenetic changes in gene expression. The Human Epigenome Consortium is developing a catalog of methylation pattern differences in the human genome which might be correlated with disease risk.
The nucleosome core is about the same size as RNA polymerase. How can RNA polymerase bind to its promoter site if it is wrapped around a nucleosome? One obvious answer is that nucleosome are not evenly distributed on chromosomal DNA, and perhaps not even found at promoter sites on the DNA. Rando et al. have studied the distribution of nucleosomes along the yeast genome. They cleaved internucleosomal DNA with nucleases leaving behind the nuclease protected-DNA. They separated the bound DNA from the nucleosome proteins, and labeled it with fluorescein. Next, total yeast DNA was isolated, fragmented, and labeled with rhodamine. They added both fluorescently labeled fragments to microarrays situated with overlapping 50 bp yeast chromosome 3 fragments. Equal red and green fluorescence at a given site on the array would arise if the DNA fragments labeled with fluorescein were protected by the nucleosome protein core particle. Low green to red fluorescence would arise if the fluorescein-labeled DNA was not protected by the nucleosome core.
From a thermodynamic viewpoint, binding affinities for the nucleosome protein core should be the same anywhere along the chromosomal DNA. This would lead to the prediction that nucleosomes would bind randomly along the DNA at all locations. leading to a constant ratio of green to red fluorescence across the array. That is, there would not be district signals from the array, but rather a smeared-out signal when the DNA was extracted from many yeast cells. The actual data showed sharp fluorescein/rhodamine signals and was consistent with fact that 70% of the nucleosomes were positioned at the same position in the DNA in different cells. Promoter sites for active genes were generally not occupied by nucleosomes. It was unclear if these sites are always free of nucleosomes or whether protein transcription factors and RNA polymerase cause the nucleosomal core proteins to slide away from the promoter sites.
Recent work suggests that positions of nucleosomes along the DNA is encoded in part by the DNA sequence itself, adding yet another "genetic code" that controls gene expressions. DNA must bend around the nucleosome core. Certain dsDNA sequences are more bendable that others, and the would be expected to have a greater chance of being involved in nucleosome complexes and less accessible for transcription. Segal et al isolated nucleosome bound DNA sequences and developed a computation model to predict which sequence of DNA would be bendable and hence be able to easily form nucleosome complexes. In other words, they calculated which DNA sequences would have high affinity for nucleosomes. They concluded that 50% of the positioning of nucleosomes can be accounted for by certain DNA sequences having higher affinity of the histone octamer. They found low nucleosome occupancy at important regulatory sites such as transcription initiation sites. Regions of the chromosome coding for tRNA and rRNA, which are highly expressed, were found to have low nucleosome occupancy.
![]() Summer 2017 A step back: DNA structure at the chromosomal level
Summer 2017 A step back: DNA structure at the chromosomal level
If access to transcription factor and RNA polymerase binding sites is critical in the control of gene expression at the level of the nucleosome, then even more fundamental would be the overall state of compaction of DNA at the the level of the chromosome itself.
Chromatin is divided into two types, euchromatin (relaxed and transcriptionally active) and heterochromatin (condensed and transcriptionally inactive). Heterochromatin is enriched in repetitive sequences and has a high degree of Lys 9 methylation on histone H3, one of the four histones that comprise the core of the nucleosome upon which DNA is wound. In addition, heterochromatin protein 1 (HP1) is recruited to the methylated histone and is thought to help in the compaction of the DNA. In such a state, transcription factors and RNA polymerase have hindered access to the their binding sites, making transcription difficult. Chromosomes that are silent are found in the compacted heterochromatin structures.
Such static images of chromatin have perhaps prevented alternative models to explain the dynamics of overall chromatin structure formation. Studies by Strom et al suggest than an alternative mechanism involving a type of phase separation called liquid-liquid demixing (discussed in Chapter 5C: Binding, Intracellular Granules and Droplets) could account for the dynamical process of chromatin compaction and hence be of critical importance in the control of transcription at the level of the chromosome.
We have seen previously that intrinsically disordered proteins are characterized by amorphous structures with repeated, often positively charged amino acids. Under the right condition, these can aggregate and “precipitate” from the solution. to form distinct liquid droplets in a proces called liquid-liquid demixing. Chromatin is a dynamic mixture of DNA and many chromosomal binding proteins. Could transcriptional processing at the chromosome level involve "phase changes" in the DNA as well?
The Drosophila HP1a protein has high intrinsic disorder and has been shown in vitro to form droplets in solution from liquid-liquid demixing processes at high protein concentration and low salt concentration. At higher temperatures (37oC), the equilibrium is shifted back to the soluble aqueous form and the drops “dissolve”. To study the process in vivo, investigators looked at early phases of heterochromatin formation as a function of time in Drosophila embryos using a green fluorescent fusion protein of HP1a (GFP-HP1a).
At the start, fluorescence from GFP-HP1a was diffuse, indicating generalized solubility in the nucleus. With time, spherical structures formed and fused with others, until prophase, the first stage of cell division when chromosomes become apparent and when the fusion protein dissociates from the chromatin. The spheres appeared again in interphase (interval between two mitotic events) but disappear during mitosis.
That the drops (small then large) are spherical suggest that they are liquid-like. Heterochromatin often appears spherical. As it gets large, it becomes less spherical which may simply reflect excess HP1a interactions with the heterochromatin. Addition of 1,6-hexanediol to Drosophila and mouse NIH3T3 significantly reduced HP1a in the drops, presumably because it disrupts weak hydrophobic interactions. If the concentration of HP1a is lowered in the cells, the appearance of another labeled chromatin protein, GFP-HP4, becomes diffuse, suggesting that HP1a is needed for drop formation.
Heterochromatin would then consist of multiple and different sized drops which could grow and retreat with time and changes in associated proteins. It would be a complex mixture of liquid drop domains and stable, non-drop structures. Their varying properties would obviously affect all aspects of chromatin structure and properties, including the control of gene transcription at the chromosomal level.