
F4. Hydrophobic Interactions: Introduction
- Page ID
- 4767

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)We have studied the role of the hydrophobic effect (involving the favorable entropic release of caged water molecules about solvent-exposed hydrophobic groups) in driving micelle and bilayer formation. Does this also drive protein folding? To explore this questions, we will study the thermodynamics of small nonpolar molecules, especially benzene, with water and ask whether the thermodynamic parameter associated with benzene solubility are similar to those associated with protein stability. If this analogy holds, anything that will promote benzene solubility will lead to increased hydrophobic amino acid side chain exposure to water and hence protein denaturation. What is the evidence to support this?
a. crystal structures: These structures show that most nonpolar side chains are buried inside a protein, which is tightly packed and which excludes water. Studies show that as the surface area of amino acid side chains increase, the free energy of transfer of amino acids from water to ethanol becomes more negative.
Figure: Transfer of amino acids from water
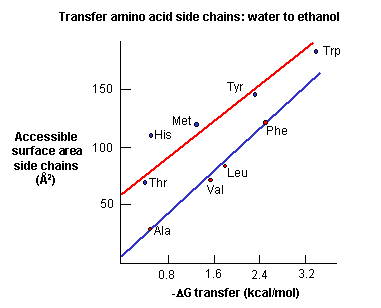
(Review free energies of transfer of hydrophobic groups in Chapter 1D: Lipids in Water - Thermodynamics )
b. low temperature denaturation of proteins - It has been observed that proteins can denature at low temperatures (less than 0oC), suggesting that nonpolar residues become more "soluble" in water at low temperatures (i.e. they move from the more hydrophobic interior of a protein to the more polar outside). Compare the solubility of nonpolar gases like CO2 or N2, which are more soluble at low temperature. As you heat solutions of nonpolar gases in water, the gases become less soluble as evidenced by bubble formation (i.e. phase separation of dissolved gases as they become more insoluble). If protein behavior is governed by this same behavior (greater solubility of nonpolar groups at low temperatures), it would suggest that proteins might denature at low temperatures (leading to increased exposure to water of the nonpolar side chains). This phenomena has been observed.
c. protein stability affected by different salt species - Over 100 years ago, Hofmeister determined the effectiveness of different cations and anions of salts to precipitate blood serum proteins in the 0.01 - 1 M concentration ranges. The series is shown below:
Cations:
NH4+ > K+ > Na+ > Li+ > Mg2+ > Ca2+ > guanidinium
Anions:
SO42- > HPO42- > acetate > citrate > Cl- > NO3- > ClO3- > I- > ClO4- > SCN-
- A salt from pairs of the first ions in these series (for example, (NH4)2SO4), when added to aqueous solutions of proteins, precipitate the native form of the protein. We must account for the fact that it precipitates the protein, and that the protein is precipitated in the native, not denatured state. More on why it precipitates proteins in a moment. The first ion in each series increases the surface tension of water (making it harder to make a cavity in the water to fit the nonpolar molecule). This decreases the solubility of nonpolar molecules. These "salt-out" nonpolar molecules, promoting not dissolution in water but aggregation followed by a phase separation. By analogy, they will stabilize the native state since the buried hydrophobic side chains would have a decreased propensity to move out into the aqueous environment.
- The last ions of the series have less affect on surface tension, and hence increase the solubility of nonpolar molecules ("salt-in"). By analogy, they will destabilize the native state since the buried hydrophobic side chains would have an increased propensity to move out into the aqueous environment.
- Hofmeister Series
Figure: Hofmeister Series

The solubility of benzene in aqueous salt solutions of this series increases from left to right, just as native protein stability decreases from left to right (i.e. the protein's nonpolar core residues become more "soluble" in water, leading to its denaturation).
d. conservation of hydrophobic core residues - These residues are highly conserved and correlated with structure.
e. Urea denatures proteins - Another additive, urea, at high concentrations is often used to denature proteins. People used to think that urea competed with the intrachain H bonds and hence unraveled the protein. The arguments above with H bonds disputes this contention since water should then denature protein. How does urea denature proteins? It has been shown that the free energy of transfer of the nonpolar amino acids into 8M urea is increasing negative as the side chains become bigger and more nonpolar.
Figure: Free energy of transfer of the nonpolar amino acids into 8 M urea
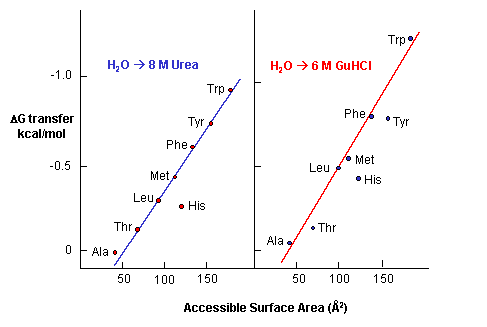
This is also true for denaturation by guanidine hydrochloride. Urea also increases the solubility of nonpolar molecules in a manner proportional to their surface area.