
23.3: Chromosome Packaging
- Page ID
- 15191


Some of the material in this chapter section comes from Chapter 8.4, Chromosomes and Chromatin, as it was important to describe it earlier in the structure/function unit. In addition, some biochemistry courses might not get to the material in a late chapter in a text. Repetition of some of the material is easier in an online textbook as well.
Introduction
Recall from Chapter 8, that within eukaryotic cells, DNA is organized into long linear structures called chromosomes, as shown in Figure \(\PageIndex{1}\). A chromosome is a deoxyribonucleic acid (DNA) molecule with part or all of the genetic material (genome) of an organism. Most eukaryotic chromosomes include packaging proteins which, aided by chaperone proteins, bind to and condense the DNA molecule to prevent it from becoming an unmanageable tangle. Before typical cell division, these chromosomes are duplicated in the process of DNA replication, providing a complete set of chromosomes for each daughter cell. The replicated arms of a chromosome are called chromatids. Before being separated into the daughter cells during mitosis, replicated chromatids are held together by a chromosomal structure called the centromere.

Eukaryotic organisms (animals, plants, fungi, and protists) store most of their DNA inside the cell nucleus as linear nuclear DNA, and some in the mitochondria as circular mitochondrial DNA or in chloroplasts as circular chloroplast DNA. In contrast, prokaryotes (bacteria and archaea) do not have organelle structures and thus, store their DNA only in a region of the cytoplasm known as the nucleoid region. Prokaryotic chromosomes consist of double–stranded circular DNA.
The genome of a cell is often significantly larger than the cell itself. For example, if the DNA from a human cell containing 46 chromosomes were stretched out in a line, it would extend more than 6 feet (2 meters)! How is it possible that the genetic information not only fits into the cell but fits into the cell nucleus? Eukaryota solves this problem by a combination of supercoiling and packaging DNA around the histone family of proteins (described below). Prokaryotes do not contain histones (with a few exceptions). Prokaryotes tend to compress their DNA using nucleoid-associated-proteins (NAPs) and supercoiling (Figure 24.3.2).
Supercoiling
DNA supercoiling refers to the over- or under-winding of a DNA strand, and is an expression of the strain on that strand, as shown in Figure \(\PageIndex{2}\). Supercoiling is important in many biological processes, such as compacting DNA, and regulating access to the genetic code. DNA supercoiling strongly affects DNA metabolism and possibly gene expression. Additionally, certain enzymes such as topoisomerases can change DNA topology to facilitate functions such as DNA replication or transcription.
In a “relaxed” double-helical segment of B-DNA, the two strands twist around the helical axis once every 10.4–10.5 base pairs of sequence. Adding or subtracting twists, as some enzymes can do, impose strain. If a DNA segment under twist strain were closed into a circle by joining its two ends and then allowed to move freely, the circular DNA would contort into a new shape, such as a simple figure-eight (Figure \(\PageIndex{2}\)). Such a contortion is a supercoil. The noun form “supercoil” is often used in the context of DNA topology.

Positively supercoiled (overwound) DNA is transiently generated during DNA replication and transcription, and, if not promptly relaxed, inhibits (regulates) these processes. The simple figure eight is the simplest supercoil and is the shape a circular DNA assumes to accommodate one too many or one too few helical twists. The two lobes of the figure- eight will appear rotated either clockwise or counterclockwise with respect to one another, depending on whether the helix is over- or underwound. For each additional helical twist being accommodated, the lobes will show one more rotation about their axis. As a general rule, the DNA of most organisms is negatively supercoiled.
Lobal contortions of a circular DNA, such as the rotation of the figure-eight lobes above, are referred to as writhe. The above example illustrates that twist and writhes are interconvertible. Supercoiling can be represented mathematically by the sum of twist and writhe (Figure \(\PageIndex{2}\). The twist is the number of helical turns in the DNA and the writhe is the number of times the double helix crosses over on itself (these are the supercoils). Extra helical twists are positive and lead to positive supercoiling, while subtractive twisting causes negative supercoiling. Many topoisomerase enzymes sense supercoiling and either generate or dissipate it as they change DNA topology.
In addition to forming supercoiled structures, circular chromosomes from bacteria have been shown to undergo the processes of catenation and knotting upon the inhibition of topoisomerase enzymes. Catenation is the process by which two circular DNA strands are linked together like chain links, whereas DNA knotting is the interlooping structures occurring within a single circular DNA structure, as shown in Figure \(\PageIndex{3}\). In vivo, the action of topoisomerase enzymes is critical to keep knots and catenoids from tangling the DNA structure. Catenanes are effectively topologically linked circular molecules

In part, because chromosomes may be very large, segments in the middle may act as if their ends are anchored. As a result, they may be unable to distribute excess twist to the rest of the chromosome or to absorb twist to recover from underwinding—the segments may become supercoiled, in other words. In response to supercoiling, they will assume an amount of writhe, just as if their ends were joined.
Supercoiled circular DNA forms two major structures; a plectoneme or a toroid, or a combination of both (Figure 24.3.2). A negatively supercoiled DNA molecule will produce either a one-start left-handed helix, the toroid, or a two-start right-handed helix with terminal loops, the plectoneme. Plectonemes are typically more common in nature, and this is the shape most bacterial plasmids will take (Figure 4.10). For larger molecules, it is common for hybrid structures to form – a loop on a toroid can extend into a plectoneme, as shown in Figure \(\PageIndex{4}\). DNA supercoiling is important for DNA packaging within all cells, and seems to also play a role in gene expression.

Topoisomerases
Topoisomerase can change the tension in supercoiled DNA. Think of how you untie a knot. It takes a lot of work sometimes, and if it's too hard, you simply cut the impediment to unknotting. Topoisomerases work by making transient breaks in the DNA before unwinding and religation. There are two main types of topoisomerases, topo I and topo II. It's very hard to describe their activities with just words and static diagrams. View the video below and you will get a great sense of what the enzymes do and how they are different.
With this background, we can now explore each enzyme in more detail. They are both targets of cancer drugs which makes them even more interesting.
Topo I enzyme relaxes DNA by nicking one stand. The dsDNA then rotates around the non-nicked strand. It unwinds new DNA and allows the condensation of chromosomes. When both DNA and RNA polymerase makes new DNA and RNA strands, respectively, they increase the supercoiling of the nucleic acid. Topoisomerases relax them. They also play a role in the regulation of gene expression by affecting gene promoters where RNA polymerase binds, with negative supercoiling enhancing transcription and positive supercoiling inhibiting it.
Figure \(\PageIndex{5}\) shows the topology of DNA and an overview of the mechanisms of Topo I and II. : DNA topology and DNA topoisomerase mechanism

Figure \(\PageIndex{5}\): DNA topology and DNA topoisomerase mechanisms. Shannon J. McKie, Keir C. Neuman, and Anthony Maxwell. Bioessays (2021). https://doi.org/10.1002/bies.202000286. Attribution 4.0 International (CC BY 4.0)
(A) Topological consequences of DNA metabolism. i) During DNA replication, strand separation leads to positive supercoiling ahead of the advancing protein machinery, and precatenane formation behind. Precatenanes form as the newly-synthesized duplexes wrap around one another, and, if not removed before completion of replication, catenated DNA molecules are formed. ii) During transcription, strand separation leads to positive supercoiling ahead of the advancing protein machinery, and negative supercoil formation behind. iii) Hemicatenanes are a possible end result of replication, in which the parental strands of the replicated duplexes remain base-paired. iv: DNA knotting can also occur as a result of DNA replication in which a DNA molecule is intramolecularly linked.
(B) Summary of topo categories and mechanism. The topos are categorized based on whether they catalyze single- (type I) or double-stranded (type II) DNA breaks. The type I topos are further subdivided to type IA, IB, and IC. Type IA form a transient covalent bond to the 5ʹ DNA phosphate and function via a strand passage mechanism. Type IB and IC form a transient covalent bond to the 3ʹ DNA phosphate and function via a controlled-rotation mechanism. Type II topos are further subdivided into type IIA and IIB. Both form a transient covalent bond to the 5ʹ DNA phosphate of both strands of the duplex and function via a strand-passage mechanism.
(C) Summary of the topological manipulations performed by DNA topoisomerases, namely relaxation of positive and negative supercoils and decatenation. Type IA topos are color-coded pink, type IB are orange, type IC are yellow, type IIA are green, and type IIB are blue. The requirement of ATP or ssDNA for activity is denoted using a red or blue circle, respectively
Topoisomerase I (Topo I):
Class I topoisomerases wrap around the DNA and cut one strand. Keeping that spot in place, the helix can spin to reduce strain caused by either over- or underwinding. After these geometric contortions, the single-stranded DNA nick is repaired and the tension is relieved. Type IA topoisomerase from E. Coli is shown in Figure \(\PageIndex{6}\).

Figure \(\PageIndex{6}\): Structure of a type IA topoisomerase. E. coli topoisomerase III is shown to illustrate the overall structure of a type IA topoisomerase and the typical toroidal fold observed in all members of this type.
(A) Diagram showing the structure of the apo-enzyme [PDB 1D6M(7)]. In the absence of DNA, the active site, found at the intersection of domains I and III (encircled), is buried.
(B) Diagram showing the structure of a complex with single-stranded DNA [PDB 1I7D (23)]. Note the movement of domains that occurs to accommodate DNA. In both diagrams, the four major domains of the protein are colored red, blue, purple, and green for domains I, II, III, and IV, respectively. The single-stranded DNA binding groove, shown circled in black, extends from domain IV to the active site. The active site residues as well as the single-stranded DNA in the complex are shown in a ball and stick representation. Dasgupta T, Ferdous S, Tse-Dinh YC. Mechanism of Type IA Topoisomerases. Molecules. 2020 Oct 17;25(20):4769. doi: 10.3390/molecules25204769. PMID: 33080770; PMCID: PMC7587558. Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
A simplified cartoon mechanism for Topo I is shown in Figure \(\PageIndex{7}\).

Figure \(\PageIndex{7}\): Diagram showing the proposed mechanism of DNA relaxation by type IA topoisomerases. The mechanism involves several transient
conformational intermediates both of the protein and the DNA. The sequence of the steps and the intermediates are hypothetical and more states are likely to be involved in the cycle. Processivity by the enzyme requires that after one relaxation event, the protein continues to another relaxation cycle without releasing the DNA. In the diagram,
the protein is shown in grey, and the DNA in red/blue. The orange dot represents the presence of the covalent protein/DNA complex. The single-stranded DNA binding groove is shown in red or yellow. Dasgupta, T et al. ibid.
A more detailed view of the domain structure and mechanism for Topo I is shown in Figure \(\PageIndex{8}\).

Figure \(\PageIndex{8}\): Type IA DNA topoisomerases. Dasgupta, T et al. ibid.
(A) Protein domain organization of Escherichia coli DNA topoisomerase IA (topo IA) and DNA topoisomerase III (topo III). Black vertical lines represent the active site tyrosines.
(B) Crystal structure of E. coli topo I bound to ssDNA (PDB: 4RUL).[20]
(C) Strand-passage mechanism for type IA topos. (1) topo binds the G-segment ssDNA region, (2) the G-segment is cleaved. (3) The topo DNA gate is opened, (4) which allows T-segment transfer through the cleaved G-strand. (5) The DNA gate is closed, (6) and the G-strand is re-ligated, changing the linking number by 1. (7) The topo can then go through another round of relaxation or dissociate from the DNA. Type IA topo (domains 1–4) is in pink, the active site tyrosine is yellow and the DNA is grey.
(D) Crystal structure of E. coli topo III bound to ssDNA (PDB: 2O54).[26]
(E) Crystal structures of human topo IIIα (blue) bound to RMI1(orange) (PDB: 4CGY),[39] and human topo IIIβ (magenta) bound to TDRD3 (green) (PDB: 5GVE).[60] For panels A, B, and C, the topo I and III domains are color-coded as follows: D1 is red, D2 is pink, D3 is yellow, D4 is orange, D5 is marine blue, D6 is purple, D7 is green, D8 is teal, and D9 is light blue
Figure \(\PageIndex{9}\) shows an interactive iCn3D model of E.Coli topoisomerase I in complex with ssDNA (4RUL).
.png?revision=1)
 Figure \(\PageIndex{9}\): E.Coli topoisomerase I in complex with ssDNA (4RUL). (Copyright; author via source).
Figure \(\PageIndex{9}\): E.Coli topoisomerase I in complex with ssDNA (4RUL). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...JxcFvds2e8p2WA
The topoisomerase is shown in gray and the single-stranded DNA is in cyan. 4 Zn2+ ions are shown. Three form Zn-finger motifs. Two of these are shown near the protein binding site for DNA. A fourth Zn is bound to a single His 566 in the structure. The active site amino acids of the enzyme, which includes residues D111, D113, Y319, and R321 from D1 and D3, are shown and labeled. Also shown are active site residues, E115, F139 and Y312. There are 4 Cys-Zn ribbon domains. The ones that interact with the ssDNA involve π-stacking, some of which are illustrated in the iCn3D model.
Class II topoisomerases (Topo II)
A series of enzymes are included in this class including DNA gyrase and Topo (IV) from prokaryotes and Topo II from eukaryotes. In eukaryotes, they help sister chromosomes separate if they get tangled during cell division. This enzyme works by: this enzyme makes a double-stranded cut, moves on the helix through the cut, and reseals the cut.
- binding the gate segment (G -segment) ds-DNA at a DNA gate where a double-stranded break is made
- binding the transport segment (T-segment) ds-DNA at the N-gate where the nucleotide ATP binds
- The T-segment DNA moves through the break in the G-segment and released the C-gate
- The G- and T-segments are reconnected.
After this, the N gate reopens to allow the process to occur again.
The domain structure of Topo IIs and the general mechanism of action are shown in Figure \(\PageIndex{10}\).

Figure \(\PageIndex{10}\): Type II DNA topoisomerases: domain organization and mechanism.
(A) Protein domain organization for the type IIA topos: E. coli DNA gyrase, E. coli DNA topoisomerase IV (topo IV), yeast DNA topoisomerase II (topo II), Methanosarcina mazei DNA topoisomerase VI (topo VI), Paenibacillus polymyxa DNA topoisomerase VIII (plasmid-borne), and Pseudomonas phage NP1 Mini-A.
(B) type II topo strand passage mechanism. (1) G-segment is bound at the DNA gate and the T-segment is captured. (2) ATP binding stimulates dimerization of the N-gate, the G-segment is cleaved and the T-segment is passed through the break. (3) The G-segment is re-ligated and T-segment exits through the C-gate. For type IIB topos, there is no C-gate so once the T-segment passes through the G-segment, it is released from the enzyme. (4) Dissociation of ADP and Pi allows N-gate opening, a scenario where the enzyme either remains bound to the G-segment, ready to capture a consecutive T-segment, or (5) dissociates from the G-segment.
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of Yeast Topoisomerase II-DNA-AMPPNP complex (4GFH) .
.png?revision=1)
Figure \(\PageIndex{11}\): Yeast Topoisomerase II-DNA-AMPPNP complex (4GFH). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...b5EHLfMwde5Dw8
The two protein chains in the homodimer are shown in magenta and cyan. The double-stranded DNA is shown with a brown backbone and CPK-colored spheres. ANP (AMPPNP), a nonhydrolyzable ATP analog, and Mg2+ ions are shown in spacefill and labeled.
The ATP binding and ATPase domain of one of the monomers (cyan for example) is adjacent to the nuclease-cutting domain of the other monomer (magenta). This requires some conformational gymnastics as the ATP binding and cleavage domain move around each other to allow the DNA strand to pass in the right direction and to reset the enzyme.
Note the circular nature of chloroplast and mitochondrial DNA, suggesting a bacterial origin for both of these organelle structures. Sequence alignments further lend support for the endosymbiotic theory, which proposes that bacteria were engulfed by early eukaryotic organisms and subsequently became symbiotic to their eukaryotic counterpart, rather than being digested.
In the cells of extant organisms, the vast majority of the proteins present in the mitochondria (numbering approximately 1500 different types in mammals) are coded for by nuclear DNA. However, sequencing of the human mitochondrial genome has revealed 16,569 base pairs encoding 13 proteins (Figure 24.3.5). Many of the mitochondrially produced proteins are required for electron transport during the production of ATP, as shown in Figure \(\PageIndex{12}\).

Histones and DNA packing
Within eukaryotic chromosomes, chromatin proteins, known as histones, compact and organize DNA. These compacting structures guide the interactions between DNA and other proteins, helping control which parts of the DNA are transcribed.
Histones are highly alkaline proteins found in eukaryotic cell nuclei that package and order the DNA into structural units called nucleosomes. They are the chief protein components of chromatin, acting as spools around which DNA winds, and playing a role in gene regulation. Without histones, the unwound DNA in chromosomes would be very long (a length-to-width ratio of more than 10 million to 1 in human DNA). For example, each human diploid cell (containing 23 pairs of chromosomes) has about 1.8 meters of DNA; wound on the histones, the diploid cell has about 90 micrometers (0.09 mm) of chromatin.
Five major families of histones exist: H1/H5, H2A, H2B, H3, and H4. Histones H2A, H2B, H3, and H4 are known as the core histones, while histones H1/H5 are known as the linker histones.
The core histones all exist as dimers, which are similar in that they all possess the histone fold domain: three alpha helices linked by two loops. It is this helical structure that allows for interaction between distinct dimers, particularly in a head-tail fashion (also called the handshake motif). The resulting four distinct dimers then come together to form one octameric nucleosome core, approximately 63 Angstroms in diameter. Around 146 base pairs (bp) of DNA wrap around this core particle 1.65 times in a left-handed super-helical turn to give a particle of around 100 Angstroms across, called a nucleosome, as shown in Figure \(\PageIndex{13}\).

The linker histone H1 binds the nucleosome at the entry and exit sites of the DNA, thus locking the DNA into place and allowing the formation of a higher order structure, as shown in Figure \(\PageIndex{14}\). The most basic such formation is the 10 nm fiber or beads on a string conformation. This involves the wrapping of DNA around nucleosomes with approximately 50 base pairs of DNA separating each pair of nucleosomes (also referred to as linker DNA).

The nucleosome contains over 120 direct protein-DNA interactions and several hundred water-mediated ones. Direct protein – DNA interactions are not spread evenly about the octamer surface but rather located at discrete sites. These are due to the formation of two types of DNA binding sites within the octamer; the α1α1 site, which uses the α1 helix from two adjacent histones, and the L1L2 site formed by the L1 and L2 loops. Salt links and hydrogen bonding between both side-chain basic and hydroxyl groups and main-chain amides with the DNA backbone phosphates form the bulk of interactions with the DNA. This is important, given that the ubiquitous distribution of nucleosomes along genomes requires it to be a non-sequence-specific DNA-binding factor. Although nucleosomes tend to prefer some DNA sequences over others, they are capable of binding practically to any sequence, which is thought to be due to the flexibility in the formation of these water-mediated interactions. In addition, non-polar interactions are made between protein side-chains and the deoxyribose groups, and an arginine side-chain intercalates into the DNA minor groove at all 14 sites where it faces the octamer surface. The distribution and strength of DNA-binding sites about the octamer surface distort the DNA within the nucleosome core. The DNA is non-uniformly bent and also contains twist defects. The twist of free B-form DNA in solution is 10.5 bp per turn. However, the overall twist of nucleosomal DNA is only 10.2 bp per turn, varying from a value of 9.4 to 10.9 bp per turn.
The histone tail extensions constitute up to 30% by mass of the histones but are not visible in the crystal structures of nucleosomes due to their high intrinsic flexibility, and have been thought to be largely unstructured (Figure 4.14). The N-terminal tails of histones H3 and H2B pass through a channel formed by the minor grooves of the two DNA strands, protruding from the DNA every 20 bp. The N-terminal tail of histone H4, on the other hand, has a region of highly basic amino acids (16-25), which, in the crystal structure, forms an interaction with the highly acidic surface region of a H2A-H2B dimer of another nucleosome, being potentially relevant for the higher-order structure of nucleosomes. This interaction is thought to occur under physiological conditions also, and suggests that acetylation of the H4 tail distorts the higher-order structure of chromatin.
Figure \(\PageIndex{15}\) shows an interactive iCn3D model of the human nucleosome (3afa). One member of each pair of histones is shown in cartoon rendering, while the other member of the pair is shown in the same color but in spacefill rendering. The structure of a human nucleosome (3afa) is shown below (H2A is shown in cyan, H2B in blue, H3 in magenta, and H4 in purple). Each strand of DNA is shown in a different shade of gray.
.png?revision=1&size=bestfit&width=285)
Figure \(\PageIndex{15}\): Human nucleosome (3afa). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...B2SwQHYDLj4BJ6
The formation of the DNA double helix represents the first-order packaging of the chromosome structure. The formation of nucleosomes represents the second level of packaging for eukaryotic chromosomes. In vitro data suggests that nucleosomes are then arranged into either a solenoid structure which consists of 6 nucleosomes linked together by the Histone H1 linker proteins or a zigzag structure that is similar to the solenoid construct, as shown in Figure \(\PageIndex{16}\). Both the solenoid and zigzag structures are approximately 30 nm in diameter. The solenoid and zigzag structures reported from in vitro data have not yet been confirmed to occur in vivo.

During interphase, each chromosome occupies a spatially limited, roughly elliptical domain which is known as a chromosome territory (CT). Each chromosome territory is comprised of higher-order chromatin units of ~1 Mb each. These units are likely built up from smaller loop domains that contain the solenoid/zigzag structural motifs. On the other hand, 1Mb domains can themselves serve as smaller units in higher-order chromatin structures.
Chromosome territories are known to be arranged radially around the nucleus. This arrangement is both cell and tissue-type specific and is also evolutionarily conserved. The radial organization of chromosome territories was shown to correlate with their gene density and size. In this case, the gene-rich chromosomes occupy interior positions, whereas larger, gene-poor chromosomes, tend to be located around the periphery. Chromosome territories are also dynamic structures, with genes able to relocate from the periphery towards the interior once they have been ‘switched on’. In other cases, genes may move in the opposite direction, or simply maintain their position. The eviction of genes from their chromosome territories into the interchromatin compartment or a neighboring chromosome territory is often accompanied by the formation of large decondensed chromatin loops.
Models describing chromosome territory arrangement
With the development of high-throughput biochemical techniques, such as 3C (chromosome conformation capture) and 4C (chromosome conformation capture-on-chip and circular chromosome conformation capture), numerous spatial interactions between neighboring chromatin territories have been described, as shown in Figure \(\PageIndex{17}\). These descriptions have been supplemented with the construction of spatial proximity maps for the entire genome (e.g., for a human lymphoblastoid cell line). Together, these observations and physical simulations have led to the proposal of various models that aim to define the structural organization of chromosome territories:

1. The chromosome territory-interchromatin compartment (CT-IC) model describes two principal compartments: chromosome territories (CTs) and an interchromatin compartment (IC). In this model, chromosome territories build up an interconnected chromatin network that is associated with an adjacent 3D space called the interchromatin compartment. The latter can be observed using both light and electron microscopy.
Within a single chromosome territory, the interphase chromosome is divided into defined regions based on the level of chromosome condensation. Here, the inner part of the interphase chromosome is comprised of more condensed chromatin domains or higher-order chromatin fibers, while a thin (<200 nm) layer of more decondensed chromatin, known as the perichromatin region, can be found around the chromosomal periphery. Functionally, the perichromatin region represents the major transcriptional compartment and is also the region where most co-transcriptional RNA splicing takes place. DNA replication and DNA repair are also predominately carried out within the perichromatin region. Finally, nascent RNA transcripts, referred to as perichromatin fibrils, are also generated in the perichromatin region. Perichromatin fibrils are then subjected to the splicing events by the factors, provided by the interchromatin compartment.
The lattice model, proposed by Dehgani et al. is based on reports that transcription also occurs within the inner, more condensed chromosome territories and not only at the interface between the interchromatin compartment and the perichromatin region. Using ESI (electron spectroscopic imaging), Dehgani et al. showed that chromatin was organized as an array of deoxyribonucleoprotein fibers of 10–30 nm in diameter. In this study, the interchromatin compartments, which are described in the CT-IC model as large channels between chromosome territories, were not apparent. Instead, chromatin fibers created a loose meshwork of chromatin throughout the nucleus that intermingled at the periphery of chromosome territories. Thus, inter- and intra-chromosomal spaces within this meshwork are essentially contiguous and together form the intra-nuclear space.
2. The interchromatin network (ICN) model predicts that intermingling chromatin fibers/loops can make both cis- (within the same chromosome) and trans- (between different chromosomes) contacts. This intermingling is uniform and makes a distinction between the chromosome territory and interchromatin compartment functionally meaningless. The advantage of the ICN model is that it permits high chromatin dynamics and diffusion-like movements. The authors propose that ongoing transcription influences the degree of intermingling between specific chromosomes by stabilizing associations between particular loci. Such interactions are likely to depend on the transcriptional activity of the loci and are therefore cell-type specific.
The cell type-specific organization of chromosome territories has been studied by measuring the volume and frequency of intermingling between heterologous chromosomes. By using 3C (chromosome conformation capture) and FISH (fluorescence in situ hybridization) to map the regions of chromosome intermingling, it was revealed that these regions contain a higher density of active genes and are enriched with markers of transcriptional activation and repression, such as activated RNAPII. By comparing the positions of the CTs in undifferentiated mouse embryonic stem (ES) cells, ES cells in early stages of differentiation, and terminally differentiated NIH3T3 cells, it was shown that fully differentiated cells had a higher enrichment of RNAPII, compared to undifferentiated or less-differentiated cells. The findings support the notion that the intermingling regions have functional significance in the nucleus and provide a basis for understanding how the radial and relative positions of chromosomal territories evolve during the process of differentiation, explaining their organization in a cell type-dependent manner.
3. The Fraser and Bickmore model emphasizes the functional importance of giant chromatin loops, which originate from chromosome territories and expand across the nuclear space to share transcription factories. In this case, both cis- and trans- oops of decondensed chromatin can be co-expressed and co-regulated by the same transcription factory.
4. The Chromatin polymer models assume a broad range of chromatin loop sizes and predict the observed distances between genomic loci and chromosome territories, as well as the probabilities of contacts being formed between given loci. These models apply physics-based approaches that highlight the importance of entropy for understanding nuclear organization. By proposing the existence of conformational chromatin ensembles with structures based on three possible homopolymer states, these models also provide alternative structures to the traditional 30 nm chromatin fiber, which has been brought into question following recent studies.
With a lack of experimental evidence to support these described models, it must be remembered that they serve only to hypothesize the structural and chemical properties of intermediate chromatin structures and to highlight unanswered questions. For example, the mechanisms that exist to control the rate and the extent of chromatin movement remain to be defined
Telomere Structures
At the ends of the linear chromosomes are specialized regions of DNA called telomeres, shown in Figure \(\PageIndex{18}\). The main function of these regions is to allow the cell to replicate chromosome ends using the enzyme telomerase, as the enzymes that normally replicate DNA cannot copy the extreme 3′ ends of chromosomes. These specialized chromosome caps also help protect the DNA ends, and stop the DNA repair systems in the cell from treating them as damage to be corrected. In human cells, telomeres are usually lengths of single-stranded DNA containing several thousand repeats of a simple TTAGGG sequence.
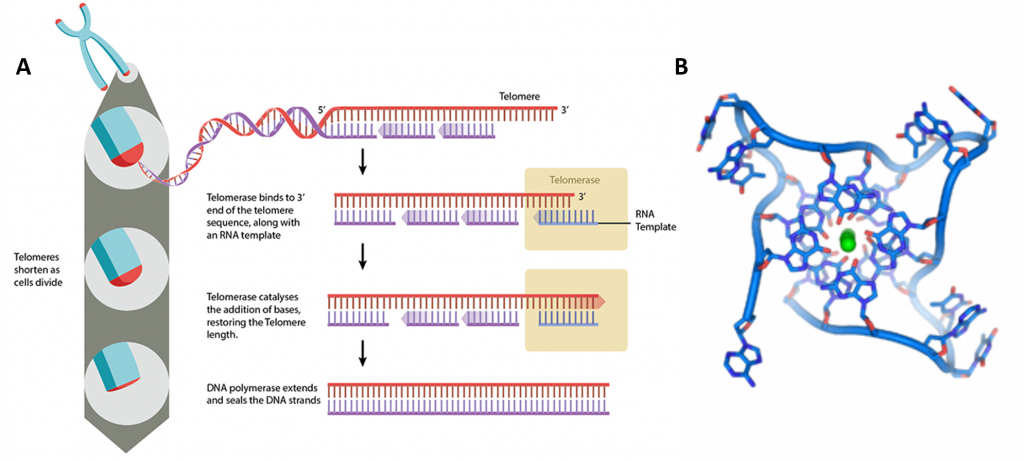
In human cells, telomeres contain 300-8000 repeats of a simple TTAGGG sequence. The repetitive TTAGGG sequences in telomeric DNA can form unique higher-order structures called quadruplexes. Figure \(\PageIndex{19}\) shows an interactive iCn3D model of parallel quadruplexes from human telomeric DNA (1KF1). The structure contains a single DNA strand (5'-AGGGTTAGGGTTAGGGTTAGGG-3') which contains four TTAGGG repeats.
.png?revision=1&size=bestfit&height=315)
Figure \(\PageIndex{19}\): A buried phenylalanine in low molecular weight protein tyrosyl phosphatase (1xww) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...y5joFHDgWJQsQ6
Rotate the model to see 3 parallel layers of quadruplexes. In each layer, 4 noncontiguous guanine bases interact with a K+ ion. Hover over the guanine bases in one layer and you will find that one layer consists of guanines 4, 10, 16, and 22, which derive from the last G in each of the repeats in the sequence of the oligomer used (5'-AGGGTTAGGGTTAGGGTTAGGG-3'). These quadruplexes certainly serve as recognition and the binding site for telomerase proteins. The guanine-rich telomere sequences which can form quadruplex may also function to stabilize chromosome ends
During DNA replication, the double-stranded DNA is unwound and DNA polymerase synthesizes new strands. However, as DNA polymerase moves in a unidirectional manner (from 5’ to 3’), only the leading strand can be replicated continuously. In the case of the lagging strand, DNA replication is discontinuous. In humans, small RNA primers attach to the lagging strand DNA, and the DNA is synthesized in small stretches of about 100-200 nucleotides, which are termed Okazaki fragments. The RNA primers are removed, and replaced with DNA and the Okazaki fragments are ligated together. At the end of the lagging strand, it is impossible to attach an RNA primer, meaning that there will be a small amount of DNA lost each time the cell divides. This ‘end replication problem’ has serious consequences for the cell as it means the DNA sequence cannot be replicated correctly, with the loss of genetic information.
To prevent this, telomeres are repeated hundreds to thousands of times at the end of the chromosomes. Each time cell division occurs, a small section of telomeric sequences is lost to the end replication problem, thereby protecting the genetic information. At some point, the telomeres become critically short. This attrition leads to cell senescence, where the cell is unable to divide, or apoptotic cell death. Telomeres are the basis for the Hayflick limit, the number of times a cell can divide before reaching senescence.
Telomeres can be restored by the enzyme telomerase, which extends telomeres length (Figure 24.3.10). Telomerase activity is found in cells that undergo regular division, such as stem cells and lymphocyte cells of the immune system. Telomeres can also be extended through the Alternative Lengthening of Telomeres (ALT) pathway. In this case, rather than being extended, telomeres are switched between chromosomes by homologous recombination. As a result of the telomere swap, one set of daughter cells will have shorter telomeres, and the other set will have longer telomeres.
A downside to telomere extension is the potential for uncontrolled cell division and cancer. Abnormally high telomerase activity has been found in the majority of cancer cells, and non-telomerase tumors often exhibit ALT pathway activation. As well as the potential for losing genetic information, cells with short telomeres are at high risk for improper chromosome recombination, which can lead to genetic instability and aneuploidy (an abnormal number of chromosomes).
These guanine-rich telomere sequences may also stabilize chromosome ends by forming structures of stacked sets of four-base units, rather than the usual base pairs found in other DNA molecules (Figure 24.3.10). Here, four guanine bases form a flat plate and these flat four-base units then stack on top of each other, to form a stable G-quadruplex structure. These structures are stabilized by hydrogen bonding between the edges of the bases and the chelation of a metal ion in the center of each four-base unit. Other structures can also be formed, with the central set of four bases coming from either a single strand folded around the bases or several different parallel strands, each contributing one base to the central structure.
In addition to these stacked structures, telomeres also form large loop structures called telomere loops or T-loops. Here, the single-stranded DNA curls around in a long circle stabilized by telomere-binding proteins. At the very end of the T-loop, the single-stranded telomere DNA is held onto a region of double-stranded DNA by the telomere strand disrupting the double-helical DNA and base pairing to one of the two strands. This triple-stranded structure is called a displacement loop or D-loop.
References
Börner, R., Kowerko, D., Miserachs, H.G., Shaffer, M., and Sigel, R.K.O. (2016) Metal ion induced heterogeneity in RNA folding studied by smFRET.Coordination Chemistry Reviews327 DOI: 10.1016/j.ccr.2016.06.002 Available at:https://www.researchgate.net/publication/303846502_Metal_ion_induced_heterogeneity_in_RNA_folding_studied_by_smFRET
Hardison, R. (2019) B-Form, A-Form, and Z-Form of DNA. Chapter in: R. Hardison’s Working with Molecular Genetics.Published by LibreTexts. Available at:https://bio.libretexts.org/Bookshelves/Genetics/Book%3A_Working_with_Molecular_Genetics_(Hardison)/Unit_I%3A_Genes%2C_Nucleic_Acids%2C_Genomes_and_Chromosomes/2%3A_Structures_of_Nucleic_Acids/2.5%3A_B-Form%2C_A-Form%2C_and_Z-Form_of_DNA
Lenglet, G., David-Cordonnier, M-H., (2010) DNA-destabilizing agents as an alternative approach for targeting DNA: Mechanisms of action and cellular consequences.Journal of Nucleic Acids2010, Article ID: 290935, DOI: 10.4061/2010/290935 Available at:https://www.hindawi.com/journals/jna/2010/290935/
Mechanobiology Institute (2018) What are chromosomes and chromosome territories? Produced by the National University of Singapore.Available at:https://www.mechanobio.info/genome-regulation/what-are-chromosomes-and-chromosome-territories/
National Human Genome Research Institute (2019) The Human Genome Project.National Institutes of Health. Available at:https://www.genome.gov/human-genome-project
Wikipedia contributors. (2019, July 8). DNA. In Wikipedia, The Free Encyclopedia. Retrieved 02:41, July 22, 2019, from https://en.Wikipedia.org/w/index.php?title=DNA&oldid=905364161
Wikipedia contributors. (2019, July 22). Chromosome. In Wikipedia, The Free Encyclopedia. Retrieved 15:18, July 23, 2019, from https://en.Wikipedia.org/w/index.php?title=Chromosome&oldid=907355235
Wikilectures. Prokaryotic Chromosomes (2017) In MediaWiki, Available at:https://www.wikilectures.eu/w/Prokaryotic_Chromosomes
Wikipedia contributors. (2019, May 15). DNA supercoil. In Wikipedia, The Free Encyclopedia. Retrieved 19:40, July 25, 2019, from https://en.Wikipedia.org/w/index.php?title=DNA_supercoil&oldid=897160342
Wikipedia contributors. (2019, July 23). Histone. In Wikipedia, The Free Encyclopedia. Retrieved 16:19, July 26, 2019, from https://en.Wikipedia.org/w/index.php?title=Histone&oldid=907472227
Wikipedia contributors. (2019, July 17). Nucleosome. In Wikipedia, The Free Encyclopedia. Retrieved 17:17, July 26, 2019, from https://en.Wikipedia.org/w/index.php?title=Nucleosome&oldid=906654745
Wikipedia contributors. (2019, July 26). Human genome. In Wikipedia, The Free Encyclopedia. Retrieved 06:12, July 27, 2019, from https://en.Wikipedia.org/w/index.php?title=Human_genome&oldid=908031878
Wikipedia contributors. (2019, July 19). Gene structure. In Wikipedia, The Free Encyclopedia. Retrieved 06:16, July 27, 2019, from https://en.Wikipedia.org/w/index.php?title=Gene_structure&oldid=906938498