
6.5: Enzymatic Reaction Mechanisms
- Page ID
- 14951


This chapter section has been written by Kristen Procko and Henry Jakubowski.
We can apply what we learned about catalysis by small molecules (e.g., acids and bases) to enzyme-catalyzed reactions. To understand the mechanism of an enzyme-catalyzed reaction, we try to alter as many variables, one at a time, and ascertain the effects of the changes on the activity of the enzyme. Kinetic methods can be used to obtain data, from which inferences about the mechanism can be made. Crystal structures of the enzyme in the presence and absence of a competitive inhibitor give abundant information about possible mechanisms. It is amazing, however, how much information about enzyme mechanism can be gained even if all you have is a blender, a stopwatch, an impure enzyme, and a few substrates and inhibiting reagents.
Introduction to Enzymes Mechanisms
Almost every chemical reaction in the biological world is catalyzed by protein enzymes. The human genome encodes for over 20,000 different proteins, thousands of which are enzymes. The total number of different enzymes in the biosphere must be staggering. Yet at the same time, all of these enzymes catalyze different sets of similar reactions. To bring order to the world of enzyme catalysis, the IUBMB has classified enzymes based on the type of chemical reactions they catalyze. There are 7 main categories as shown in the expandable Table \(\PageIndex{1}\) below. Each reaction type is given a four digit Enzyme Commission number. For example, alcohol dehydrogenase, the enzyme that catalyzes the oxidation of ethanol, a primary alcohol, to acetaldehyde using an oxidizing agent called NAD+, is given the enzyme commission number EC 1.1.1.1. Other enzymes that oxidize primary alcohols to aldehydes or secondary alcohols to ketones are also give the same EC number.
| Class | Subclass | Type | Description |
| EC 1 | [+] | Oxidoreductases | redox reactions |
| EC 2 | [+] | Transferases | transfer/exchange of group from one molecule to another |
| EC 3 | [+] | Hydrolases | hydrolysis reactions |
| EC 4 | [+] | Lyases | elimination forming double bond |
| EC 5 | [+] | Isomerases | conversions of geometric, stereo- or constitutive isomers |
| EC 6 | [+] | Ligases | condensation of two molecules into one |
| EC 7 | [+] | Translocases | movement of species across a semipermeable membrane |
Table \(\PageIndex{1}\): ExplorEnz database that for the curation and dissemination of the International Union of Biochemistry and Molecular Biology (IUBMB) Enzyme Nomenclature. (Source: https://academic.oup.com/nar/article...1/D593/1000297)
Enzymes with the same or similar EC numbers probably have similar reaction mechanisms. Throughout this book, we will explore the reaction mechanisms of many enzymes, but we can't and shouldn't explore all of them. You can take your acquired understanding of the reaction mechanism for key representative enzymes and apply them to others. Of course, experimental evidence is needed to validate a given mechanism. In this chapter section, we will focus on the mechanisms of a few transferases (EC2) and hydrolases (EC3) as prototypical examples.
Arrow Pushing Conventions in Biochemical Mechanisms
The rules for electron pushing in biochemical mechanisms mirror those from organic chemistry. However, because of the length of some biochemical mechanisms, abbreviated mechanisms are often accepted and will be presented in the literature and textbooks. The aim of this section is to review arrow pushing by presenting some simple biochemical mechanisms, and to familiarize you with acceptable alternative ways to show arrow pushing.
Class I Methyltransferases
Coenzymes are organic molecules that participate in some enzyme-catalyzed reactions (see Section 6.8 for a detailed discussion). Often, these "enzyme helpers" impart reactivity that an enzyme would not have on its own. We'll begin our investigation with a mechanism catalyzed by class I methyltransferases, which bind the coenzyme S-adenosylmethionine (SAM). SAM itself is formed from the reaction of methionine with ATP, resulting in the positively charged sulfur shown in Figure \(\PageIndex{1}\). The blue methyl group attached to the sulfur is very electrophilic due to the sulfur cation, and is transferred to a nucleophilic substrate.
The reaction in Figure \(\PageIndex{1}\) shows the SAM-promoted conversion of norepinephrine to epinephrine. It is catalyzed by the enzyme phenylethanolamine N-methyltransferase (EC: 2.1.1.28). The nucleophilic nitrogen atom in norepinephrine is brought into proximity of the electrophilic methyl group when it binds to the enzyme. In an SN2 reaction, nitrogen attacks, and we show a second arrow to keep track of the electrons from the carbon-sulfur bond, becoming a lone pair on the sulfur atom.
An amine has a pKa close to 30, but a protonated amine has a pKa around 8–10. Specifically, the conjugate acid of epinephrine has a pKa near 8. Therefore, following the attack step, a protonated amine is a reasonable product to show at physiological pH (near 7.4). However, biochemical products are often shown uncharged when depicting an overall reaction. Therefore, a general base can be shown deprotonating the ammonium ion. The general base could be an amino acid side chain, or a general base within the buffer components contained in a cell; we will represent the general base as B: here.

Figure \(\PageIndex{1}\): Class I methyltransferase mechanism with substrate norepinephrine
To simplify the mechanism in Figure \(\PageIndex{1}\), biochemists may abbreviate the arrow pushing steps. As shown by the one-step deprotonation in Figure \(\PageIndex{2}\), the general base deprotates the amine. The electrons from that bond are used to attack, and deprotonated epinephrine is produced in a single step with that arrow pushing. It is important to keep in mind that an anionic nitrogen is not produced in the reaction mechanism. With pKa values near 30, most amines are only deprotonated in practice using very strong organic bases, such as butyllithiums.
Because of this, the authors prefer the alternative arrow pushing mechanism shown at the bottom of Figure \(\PageIndex{2}\). Partial bond formation between the nitrogen and the methyl group must occur before a biological general base is able to deprotonate norepinephrine, so showing the arrow coming from the lone pair on nitrogen is perhaps a better depiction of the biological reality. The arrow pushing shown in each of the methyltransferase mechanisms is acceptable, and you may see each of these in different contexts.

Figure \(\PageIndex{2}\): Class I methyltransferase mechanism alternative arrow pushing
Kinases
Transfer of a phosphate group is common in biochemistry. The phosphate group is often transferred from adenosine triphosphate (ATP) via kinase enzymes. The following excerpt from Chemistry LibreTexts describes the phosphate transfer mechanism:
In a phosphate transfer reaction, a phosphate group is transferred from a phosphate group donor molecule to a phosphate group acceptor molecule as shown in Figure \(\PageIndex{3}\).
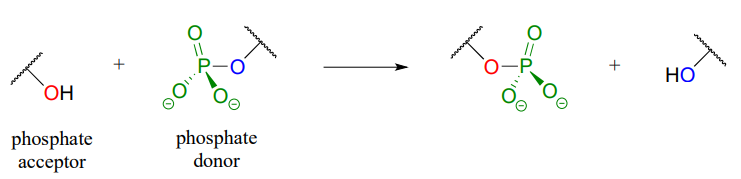
Figure \(\PageIndex{3}\): Phosphate transfer to an acceptor
A very important aspect of biological phosphate transfer reactions is that the electrophilicity of the phosphorus atom is usually enhanced by the Lewis acid (electron-accepting) effect of one or more magnesium ions. Phosphate transfer enzymes generally contain a Mg2+ ion bound in the active site in a position where it can interact with non-bridging phosphate oxygens on the substrate (Figure \(\PageIndex{4}\)). The magnesium ion pulls electron density away from the phosphorus atom, making it more electrophilic.
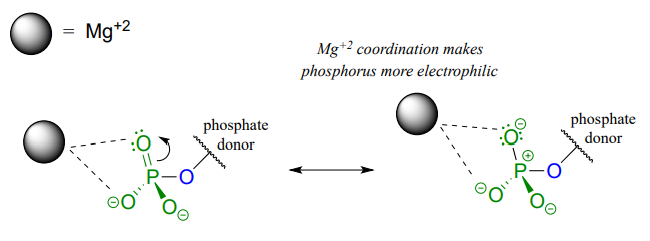
Figure \(\PageIndex{4}\): Phosphate interaction with magnesium ion
Without this metal ion interaction, a phosphate is actually a poor electrophile, as the negatively-charged oxygens shield the phosphorus center from attack by a nucleophile.
Note: For the sake of simplicity and clarity, we may omit the magnesium ion or other active site groups interacting with phosphate oxygens in some of the figures that follow—but it is important to keep in mind that these interactions play an integral role in phosphate transfer reactions.
Mechanistically speaking, a phosphate transfer reaction at a phosphorus center can be thought of as much like a SN2 reaction at a carbon center. Just like in an SN2 reaction, the nucleophile in a phosphoryl transfer approaches the electrophilic center opposite the leaving group, from the backside as shown in Figure \(\PageIndex{5}\).
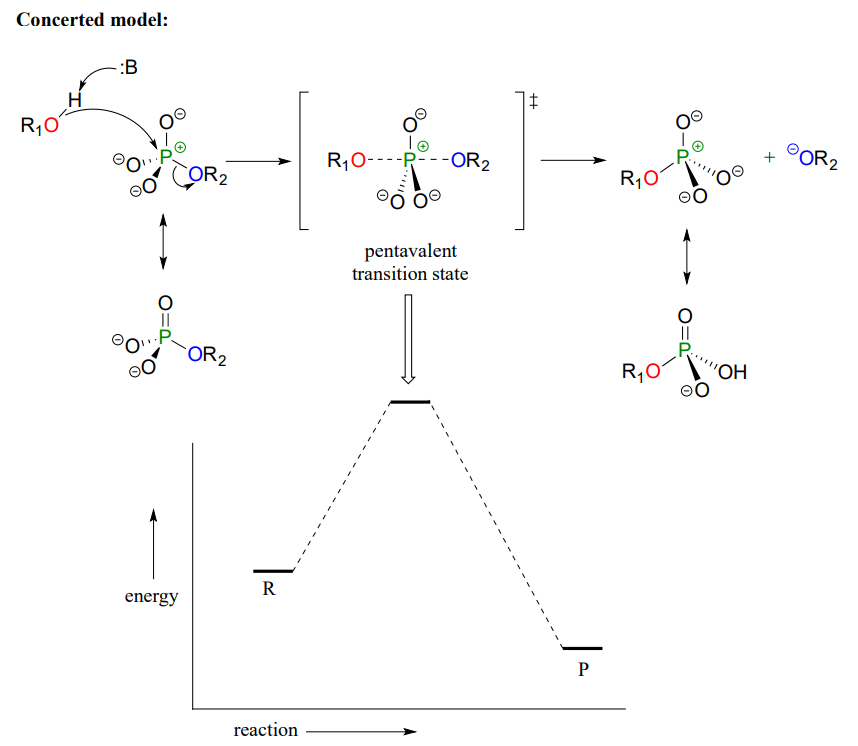
Figure \(\PageIndex{5}\): Phosphate transfer mechanism and energy diagram
As the nucleophile gets closer and the leaving group begins its departure, the bonding geometry at the phosphorus atom changes from tetrahedral to trigonal bipyramidal at the pentavalent (5-bond) transition state (Figure \(\PageIndex{6}\)). As the phosphorus-nucleophile bond gets shorter and the phosphorus-leaving group bond grows longer, the bonding picture around the phosphorus atom returns to its original tetrahedral state, but the stereochemical configuration has been 'flipped', or inverted.
In the trigonal bipyramidal transition state, the five substituents are not equivalent: the three non-bridging oxygens are said to be equatorial (forming the base of a trigonal bipyramid), while the nucleophile and the leaving group are said to be apical (occupying the tips of the two pyramids).
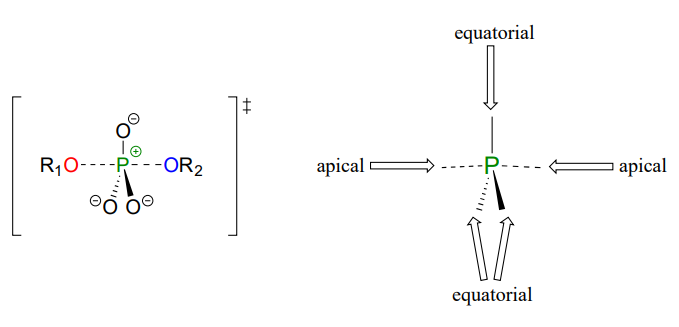
Figure \(\PageIndex{6}\): Transition state of the phosphate transfer reaction
Although stereochemical inversion in phosphoryl transfer reactions is predicted by theory, the fact that phosphoryl groups are achiral made it impossible to observe the phenomenon directly until 1978, when a group of researchers was able to synthesize organic phosphate esters in which stable oxygen isotopes 17O and 18O were specifically incorporated (Figure \(\PageIndex{7}\)). This created a chiral phosphate center.
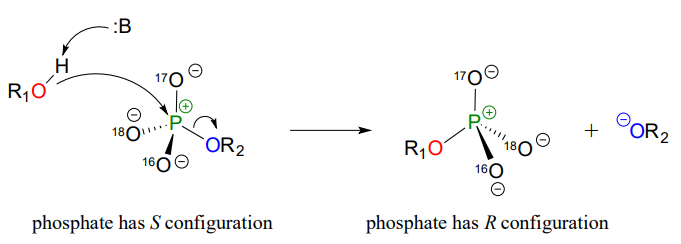
Figure \(\PageIndex{7}\): Inversion of stereochemistry in the phosphate transfer reaction
Subsequent experiments with phosphoryl transfer-catalyzing enzymes confirmed that these reactions proceed with stereochemical inversion. (Nature 1978 275, 564; Ann Rev Biochem 1980 49, 877). (The previous excerpt has been adapted from Chemistry LibreTexts.)
We should note that, although the charge-separated resonance form shown above contributes to the structure and therefore contributes to the resonance hybrid, the phosphate group is almost always shown with a double bonded oxygen atom to the phosphorous. Note that we have also depicted the mechanism below with our preferred biochemical arrow pushing, where the attack of oxygen at phosphorous facilitates the deprotonation of the alcohol (R1OH) by a general base.

Figure \(\PageIndex{8}\): Common arrow pushing for phosphate transfer
With a formal charge of negative four, ATP would be an extremely poor electrophile. Therefore, the phosphoryl transfer from ATP, shown for hexokinase (EC 2.7.1.1) in Figure \(\PageIndex{9}\), requires binding of a magnesium ion with ATP. The magnesium ion partially neutralizes the negative charge, allowing for nucleophilic attack by the oxygen atom of glucose.

Figure \(\PageIndex{9}\): Hexokinase mechanism with phosphate transfer from ATP
A crystal structure of the enzyme was solved with the substrate, glucose-6-phosphate, bound. The magnesium cofactor also occupies the active site, along with a non-hydrolyzable analog of ATP. In the substrate analog, called ANP, the oxygen of the terminal phosphoanhydride bond of ATP is replaced with a nitrogen atom, and allows us to view probable interactions that occur between the enzyme of the catalytic complex. In the iCn3D image shown in Figure \(\PageIndex{10}\), ANP and glucose are bound in the active site. The magnesium ion is shown in green.
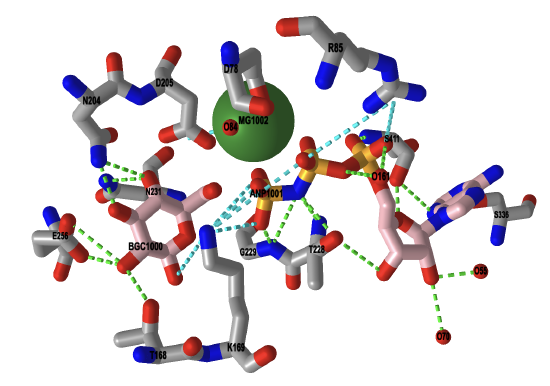
 Figure \(\PageIndex{10}\): Interactive iCn3D image of the catalytic complex of human glucokinase (a hexokinase isoform, 3FGU). (Copyright; author via source).
Figure \(\PageIndex{10}\): Interactive iCn3D image of the catalytic complex of human glucokinase (a hexokinase isoform, 3FGU). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?EajeSCpF8GM16HYq5
Elucidation of Reaction Mechanisms Using Kinetic Data
To to this point, we've presented mechanisms with the support of PDB structures alone. However, much was known about enzyme mechanisms prior to ready access to crystal structures in the Protein Data Bank. Systematically, the kineticists, medicinal chemists and molecular biologists (i.e., a well trained chemist) can change:
- the substrate - for example, changing the leaving group or substituents of a hydrolyzable substrate;
- the pH or ionic strength - which can give data about general acids/bases in the active site;
- the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis;
- the solvent - an odd idea on the surface but it leads to new insights into enzyme catalysis.
For the following enzymes, we will concentrate on reaction mechanisms based on a mix of structural data, alongside kinetic data to hypothesize a reaction mechanism consistent with the findings. Even with lots of data, there are often different proposed mechanisms for a given reaction. Kinetic data is vital as it can help to determine:
- the order of binding/dissociation of substrates and products;
- the rate constants for individual steps;
- and clues to the nature of catalytic groups found in the enzyme.
Chymotrypsin and Other Endoproteases
Chymotrypsin (EC 3.4.21.1), an endoprotease, cleaves an internal peptide bond after aromatic side chains by hydrolysis. It also cleaves small ester and amide substrates after aromatic residues. As an example, in Figure \(\PageIndex{11}\), cleavage occurs on the C-terminal side of the tyrosine residue, giving two peptide fragments.

Figure \(\PageIndex{11}\): Chymotrypsin cleavage of an example peptide substrate
Chymotrypsin has a similar mechanism to a multitude of other proteases that used the same catalytic triad, Ser 195, Asp 102 and His 95, so we'll study it significant detail. In determining the mechanism of an enzyme, you have to change an experimental variable and see how catalytic activity changes. What can be changed? Turns out everything including the solvent! Let's explore these changes and how they affect chymotrypsin activity.
- Changing the substrate (for example changing the leaving group or acyl substituents of a hydrolyzable substrate):
In the lab, it's easier to study the enzyme using small substrate mimics of a protein than to use a full protein substrate. The mimics include both esters and amides. Data from the cleavage of small amide and ester substrates shown in Figure \(\PageIndex{12}\) suggest that a covalent intermediate is formed during chymotrypsin catalyzed cleavage.

Table \(\PageIndex{2}\) below shows kinetic data for the cleavage of these substrates.
| Chymotrypsin substrate cleavage, 25 oC, pH 7.9 | |||
| kinetic constants | Acetyl-Tyr-Gly-amide | Acetyl-Tyr-O Ethylester | Ester/Amide |
| kcat (s-1) | 0.50 | 193 | 390 |
| Km (M) | 0.023 | 0.0007 | 0.03 |
| kcat/Km (M-1s-1) | 22 | 280,000 | 12,700 |
| Kinetic constants for chymotrypsin cleavage of N-acetyl-L-Trp Derivatives - N-acetyl-L-Trp-X | ||
| X | kcat (s-1) | Km x 103 (M) |
| -OCH2CH3 | 27 | 0.097 |
| -OCH3 | 28 | 0.095 |
| -p-nitrophenol | 31 | 0.002 |
| -NH2 | 0.026 | 7.3 |
Table \(\PageIndex{2}\): Cleavage of peptides and ethylester substrate analogs by chymotrypsin
Here's how these data can be interpreted.
- The kcat and kcat/Km are larger and the Km smaller for ester substrates compared to amide substrates, suggesting that amides are more difficult to hydrolyze (Table 2 above). This is expected given the poorer leaving group of the amide.
- The kcat for the hydrolysis of ester substrates doesn't depend on the nature of the leaving group (i.e., whether it is a poorer leaving group such as methoxy or a better leaving group such as p-nitrophenolate) suggesting that this step is not the rate limiting step for ester cleavage. Without the enzyme, p-nitrophenyl esters are cleaved much more rapidly than methyl esters. Therefore deacylation must be rate limiting. But deacylation of what? If water was the nucleophile, release of the leaving group would result in both products, the free carboxyl group and the amine being formed simultaneously. Since they are not released simultaneously, this suggests an acyl-enzyme covalent intermediate.
When the acyl end of the ester substrate is changed, without changing the leaving group (a p-nitrophenyl group), a covalent intermediate can be trapped. Specifically, the deacylation of a trimethyacetyl group is much slower than an acetyl group. It is so slow that a 14C-labeled trimethylacetyl-labeled chymotrypsin intermediate can be isolated after incubation of chymotrypsin with 14C-labeled p-nitrophenyltrimethylacetate using gel filtration chromatography.
We have seen a kinetic mechanism previously consistent with these ideas before. The data suggest a mechanism based on the chemical equations shown in Figure \(\PageIndex{13}\):
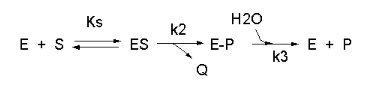
In this reaction, a substrate S might interact with E to form a complex, which then is cleaved to products P and Q. Q is released from the enzyme, but P might stay covalently attached, until it is expelled. This conforms exactly to the mechanism described above. For chymotrypsin-catalyzed cleavage, the step characterized by k2 is the acylation step. The step characterized by k3 is the deacylation step in which water attacks the acyl enzyme to release product P (free phosphate in Lab 5). The mathematical equation for this reaction is shown below (without derivation)
\begin{equation}
\mathrm{v}_{0}=\frac{\left(\frac{\mathrm{k}_{2} \mathrm{k}_{3}}{\mathrm{k}_{2}+\mathrm{k}_{3}}\right) \mathrm{E}_{0} \mathrm{~S}}{\mathrm{~K}_{\mathrm{S}}\left(\frac{\mathrm{k}_{3}}{\mathrm{k}_{2}+\mathrm{k}_{3}}\right)+\mathrm{S}}
\end{equation}
For hydrolysis of ester substrates, which have better leaving groups compared to amides, deacylation is rate limiting, ( k3<<k2). For amide hydrolysis, as mentioned above, acylation can be rate-limiting (k2<<k3). From this, equation 6.5.1 can be simplified as shown in Table \(\PageIndex{3}\) below for ester and amide hydrolysis.
| Ester hydrolysis (deacylation rate limiting, k3 << k2) | Amide hydrolysis (deacylation rate limiting, k2 << k3) |
| \begin{equation} \mathrm{v}_{0}=\frac{\mathrm{k}_{3} \mathrm{E}_{0} \mathrm{~S}}{\mathrm{~K}_{\mathrm{S}}\left(\frac{\mathrm{k}_{3}}{\mathrm{k}_{2}}\right)+\mathrm{S}} \end{equation} |
\begin{equation} \mathrm{v}_{0}=\frac{\mathrm{k}_{2} \mathrm{E}_{0} \mathrm{~S}}{\mathrm{~K}_{\mathrm{S}}+\mathrm{S}} \end{equation} |
| \begin{equation} V_{M}=k_{3} E_{0} \end{equation} |
\begin{equation} \mathrm{V}_{\mathrm{M}}=\mathrm{k}_{2} \mathrm{E}_{0} \end{equation} |
| \begin{equation} \mathrm{K}_{\mathrm{M}}=\mathrm{K}_{\mathrm{S}}\left(\frac{\mathrm{k}_{3}}{\mathrm{k}_{2}}\right) \end{equation} |
\begin{equation} \mathrm{K}_{\mathrm{M}}=\mathrm{K}_{\mathrm{S}} \end{equation} |
Table \(\PageIndex{3}\): Simplification of equation 6.5.1
Just as we saw before for the rapid equilibrium assumption (when ES falls apart to E + S more quickly than it goes to product, Chapter 6.3), KM = Ks in the case of amide hydrolysis.
This reaction with two reactants (bi) and two products (bi), a covalent enzyme intermediate, and with the second reactant binding after one the first product Q is released, is called a BiBi Ping Pong reaction. If k2 >> k3, an immediate and fast burst or release of product Q happens, followed by a slow release of P since the covalent E-P complex reacts with the second reactant with a small rate constant k3. When doing initial rate Michaelis-Menten kinetics, the initial velocity of Q formation, v0 Q is not (dQ/dt)t=0, but the slower constant rate after the burst phase, which is determined by k3, the rate of cleavage of the E-P intermediate.
The VCell computational model below shows the reaction BiBi Ping Pong reaction for the reaction involving an enzyme-P covalent intermediate. The burst phases in Q is clearly seen if you rescale the graph as described below.
 MODEL
MODEL
BiBi-Ping Pong_Covalent Intermediate Irreversible reaction
Vcell reaction diagram (1-way arrows defined as reversible in actual mathematical model) and chemical equation
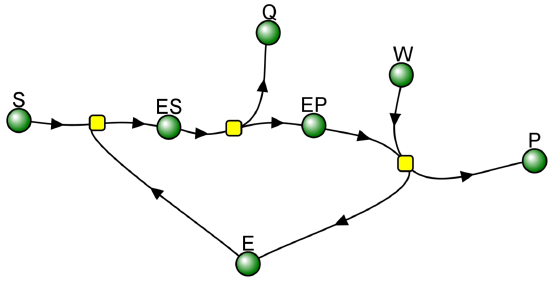
Yellow dots: Reaction Nodes (R1, R2 and R3 left to right)

Reaction made irreversible since kr2 = 0, kr3 = 0.
Initial parameter values:
- S0 = 100, E0 = 1, W (water in a hydrolysis reaction) = 50 and fixed throughout
- k1f = 5, k1r = 1, k2f = 0.6,
- k2f = 50, k2r = 0
- k3f = 0.05, k3f = 0
Select Load [model name] below
Select Start to begin the simulation.
Select Plot to change Y axis min/max, then Reset and Play | Select Slider to change which constants are displayed | Select About for software information.
To see the burst phase for reaction, change the time and parameters to these values:
- set Run time to 0.3
- Select Plot then Update Y axis max to 2
- Click Edit Plot Species and check just P and Q
- reset
Time course model made using Virtual Cell (Vcell), The Center for Cell Analysis & Modeling, at UConn Health. Funded by NIH/NIGMS (R24 GM137787); Web simulation software (miniSidewinder) from Bartholomew Jardine and Herbert M. Sauro, University of Washington. Funded by NIH/NIGMS (RO1-GM123032-04)
The burst phase is seen with ester hydrolysis as described above when k2 >> k3.
- Changing the pH or ionic strength - which can give data about general acids/bases in the active site:
- a graph of kcat as a function of pH indicates that a group of pKa of approximately 6 must be deprotonated to express activity (i.e., Vmax/2 is at about pH 6). This suggests that an active site histidine is necessary, which, if it must be deprotonated to express activity, must be acting as a general base.
- a graph of kcat/Km shows a bell-shaped curve indicating the necessity of a deprotonated side chain with a pKa of about 6 (i.e., the same His above) and a group which must be protonated with a pKa of about 10. This turns out to be an N terminal Ile in chymotrypsin, which must be protonated to form a stabilizing salt bridge in the protein. Note: This N-terminal Ile is actually at the 16 position in the inactive precursor of chymotrypsin (called chymotrypsinogen); upon activation of chymotrypsinogen loses the first 15 amino acids by selective proteolysis.
(Note: The PKAD is a database of experimentally measured pKa values of ionizable groups in proteins. It is searchable by the PDB ID.)
- Changing the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis:
Here are some specific examples.
- Modification of chymotrypsin (and many other proteases) with diisopropylphosphofluoridate (DIPF) modifies only one (Ser 195) of many serines in the protein, suggesting that it is hypernucleophilic and probably the amino acid that attacks the carbonyl C in the substrate, forming the acyl-intermediate. This reaction is illustrated in Figure \(\PageIndex{13}\). The figure also shows analagous molecules used in common insecticides, which act through a similar mechanism.

- Modification of the enzyme with tos-L-Phe-chloromethyl ketone inactives the enzyme with a 1:1 stoichiometry which results in a modified His, as shown in Figure \(\PageIndex{14}\).

- Comparison of the primary sequence of many proteases show that three residues are invariant: a Ser, a His, and an Asp residue.
- Site-specific mutagenesis show that if Ser 195 is changed to Ala 195, the enzymatic activity is almost reduced to background. The strongly suggests that Ser 195 is an active site nucleophile.
D. Changing the solvent. Yes indeed you can take chymotrypsin and show that it is active in anhydrous organic solvents. Surely this is impossible you say! It is true and we will explore it at the end of chapter since its challenging enough to understand chymotrypsin activity in aqueous solution. No new chemistry is needed, just a change in what your mind can conceptualize.
The Chymotrypsin Arrow-Pushing Mechanism
The chymotrypsin mechanism will be presented to explore the different types of acceptable arrow pushing one can show for this nucleophilic acyl substitution mechanism. In the mechanism, only the peptide bond will be shown. It is only necessary to focus on this small portion of the molecule shown in Figure \(\PageIndex{15}\) to show the arrow pushing.

Figure \(\PageIndex{15}\): Abbreviated chymotrypsin peptide cleavage reaction
The active site of chymotrypsin contains a catalytic triad, three amino acids working together to carry out the reaction that cleaves the peptide bond. The amino acids involved are the aspartate, histidine, and serine residues mentioned earlier (Figure \(\PageIndex{16}\)).

Figure \(\PageIndex{16}\): Serine protease catalytic triad
The deprotonated aspartate side chain acts to increase histidine’s basicity, allowing it to accept a proton from serine in the catalytic mechanism. In some mechanisms, Asp is shown accepting a proton from histidine; however, simplified arrow pushing can be shown without Asp acting as a proton acceptor, and that is how the mechanism will be represented here.
In the first stage of the mechanism, histidine deprotonates serine, which acts as a nucleophile and attacks the partially electropositive carbon atom of the carbonyl functional group (Figure \(\PageIndex{17}\)). In the simplest form of arrow pushing that can be shown, histidine deprotonates the nucleophilic using the lone pair on nitrogen, and the electrons from the hydrogen-oxygen bond are shown attacking. The carbonyl double bond breaks, shifting the electrons onto oxygen.
This forms a tetrahedral sp3 hybridized carbon atom from the sp2 hybridized carbonyl group, and is therefore called a tetrahedral intermediate.

Figure \(\PageIndex{17}\): First stage of the chymotrypsin mechanism
Using the simple arrow pushing model again, the electrons from the negatively-charged tetrahedral intermediate reform a double bond, kicking out the amine leaving group, which accepts an H+ from protonated histidine, neutralizing the charge on histidine. The arrow from the N-H bond neutralizes the positive charge on histidine; please note that this arrow is important to show. In all deprotonations, it is a convention to show electrons from a breaking bond becoming a lone pair on the atom receiving them.
Another acceptable form of arrow pushing for this stage of the reaction requires more arrows, but better depicts how the enzyme is interacting with the substrate in the active site (Figure \(\PageIndex{18}\). Because an active site often uses entropy reduction, bringing substrates close together in a reactive orientation, the lone pairs on heteroatoms are already interacting favorably to form the new bond. Additionally, in the first mechanism, it almost appears that a serine alkoxide attacks the carbon of the peptide bond, and that R-NH- (a poor leaving group) departs the molecule, picking up a proton after the bond breaks.
Therefore, in this second mechanism option, these subtleties are considered. In the first step, the proton on serine is deprotonated by histidine, and those electrons are pushed toward the serine oxygen atom. The serine oxygen is positioned close to the carbonyl carbon of the amide bond, and an arrow originating from the serine lone pair depicts the attack. This type of arrow pushing implies the attack and deprotonation steps are happening in concert.

Figure \(\PageIndex{18}\): Alternate arrow pushing for the first stage of the chymotrypsin mechanism
In the second step, the electrons that push down from the negatively charged oxygen atom break the N-H bond, becoming a lone pair on oxygen. Simultaneously, the lone pair already present on nitrogen is shown deprotonating histidine.
It is important to note that, because biochemical mechanisms are often lengthy, they may be shown with the tetrahedral intermediate omitted. This arrow pushing for the first step, which may be shown using either convention described above, shows the serine oxygen attacking and the amine leaving group departure in one step, as shown in Figure \(\PageIndex{19}\) below. Note that this abbreviated style of arrow pushing, which does not show the tetrahedral intermediate, often uses generic acids and bases, so it does not keep track of protonation states, or account for the amino acid residues performing protonation and deprotonation steps (which is quite important in the chymotrypsin mechanism).

Figure \(\PageIndex{19}\): Abbreviated arrow pushing for the first stage of the chymotrypsin mechanism
The covalent intermediate must be released from the enzyme in order for chymotrypsin to catalyze another reaction. This second nucleophilic acyl substitution also proceeds through a tetrahedral intermediate; this time, water is the nucleophile, as shown in Figure \(\PageIndex{20}\).

Figure \(\PageIndex{20}\): Second stage of the chymotrypsin mechanism
Now, that we've seen the steps in detail, let's put all this together to show the full mechanism for serine protease cleavage of protein, shown in Figure \(\PageIndex{21}\).

Here a summary of what the Figure \(\PageIndex{21}\) mechanism shows:
- The deprotonated His 57 acts as a general base to abstract a proton from Ser 195, enhancing its nucleophilicity as it attacks the electrophilic C of the amide or ester link, creating the oxyanion tetrahedral intermediate. Asp 102 acts electrostatically to stabilize the positive charge on the His.
- The oxyanion collapses back to form a double bond between the O and the original carbonyl C, with the amine product as the leaving group. The protonated His 57 acts as a general acid donating a proton to the amine leaving group, regenerating the unprotonated His 57.
- The mechanism repeats itself, only now with water as the nucleophile, which attacks the acyl-enzyme intermediate, to form the tetrahedral intermediate.
- The intermediate collapses again, releasing the E-SerO- as the leaving group which gets reprotonated by His 57, regenerating both His 57 and Ser 195 in the normal protonation state. The enzyme is now ready for another catalytic round of activity.
- The mechanism for the first nucleophilic attack (by Ser) is the same as for the second (by water). The reverse mechanism of condensation of two peptide would be the reverse of the above mechanism, and is an example of the principle of microscopic reversibility.
In short, many of the catalytic mechanisms we encountered previously are at play in chymotrypsin catalysis. These include nucleophilic catalysis (with the Ser 195 forming a covalent intermediate with the substrates), general acid/base catalysis with His 57, and loosely, electrostatic catalysis with Asp 102 stabilizing not the transition state or intermediate, but the protonated form of His 57. An important point to note is that His, as a general acid and base catalyst, not only stabilizes developing charges in the transition state, but also provides a path for proton transfer, without which, reactions would have difficulty in proceeding.
One final mechanism is at work. The enzyme does indeed bind the transition state more tightly than the substrate. Crystal structures with poor "pseudo"-substrates that get trapped as partial tetrahedrally-distorted substrates of the enzyme and with inhibitors show that the oxyanion intermediate, and hence presumably the TS, can form H-bonds with the amide H (from the main chain) of Gly 193 and Ser 195. These cannot be made to the trigonal, sp2 hybridized substrate. In the enzyme alone, the hole into which the oxyanion intermediate and TS would be placed is not occupied. This oxyanion hole is occupied in the tetrahedral intermediate.
A crystal structure of a relative of chymotrypsin, trypsin, which cleaves after positively charged lysine and arginine side chains, has been determined with a bound transition state analog inhibitor. The transition state inhibitor is t-butoxy-Ala-Val-boro-Lys methyl ester shown in Figure \(\PageIndex{22}\).

Recall from introductory chemistry that neutral boron compounds like BH3 and BF3 are trigonal planar (sp2) and electron deficient. Although the boron is not charged, it has a significant partial positive charge (δ+) so it is electrophilic. The nucleophilic oxygen of Ser 195 can then attack the boron to form a tetrahedral intermediate. This intermediate is not an oxyanion, but one of the attached oxygens with a δ- charge occupies the oxyanion hole.
Figure \(\PageIndex{23}\) show the active site group in trypsin interacting with part of the transition state analog (1BTZ). The serine 195 side chain O is covalently attached to the boron, so the boron is now tetrahedral (sp3).

The yellow dotted lines show hydrogen bonding between the backbone amide hydrogens of Ser 195 and Gly 193 with the methoxy oxygen of the now tetrahedral borate transition state analog inhibitor. The boron atom is the yellow/orange sp3 atom connected to 3 oxygen (red) atoms and one carbon (cyan) atom. Normally, the oxyanion O- from the tetrahedral intermediate in amide or ester cleavage would occupy the oxyanion hole.
Figure \(\PageIndex{24}\) shows an interactive iCn3D model of the active site of the phenylethane boronic acid (PBA) complex of alpha-chymotrypsin (6cha).
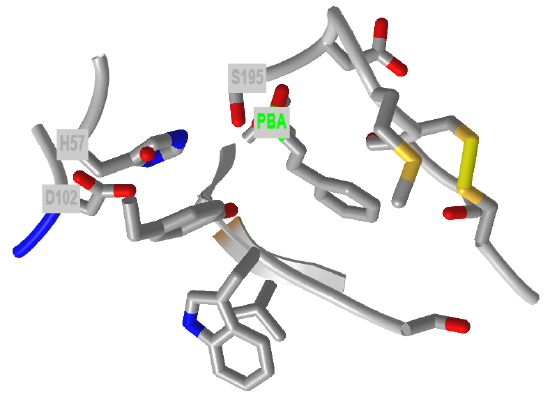
Figure \(\PageIndex{24}\): Active site of the phenylethane boronic acid (PBA) complex of alpha-chymotrypsin (6cha). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...qXUxbrakurYmx6
Many enzymes have active site serines which act as nucleophilic catalysts in nucleophilic substitution reactions (usually hydrolysis). One such enzyme is acetylcholinesterase, which cleaves the neurotransmitter acetylcholine in the synapse of the neuromuscular junction (Figure \(\PageIndex{25}\)).

Figure \(\PageIndex{25}\): Reaction catalyzed by acetylcholinesterase
The neurotransmitter leads to muscle contraction when it binds its receptor on the muscle cell surface. The transmitter must not reside too long in the synapse, otherwise muscle contraction will continue in an uncontrolled fashion. To prevent this, a hydrolytic enzyme, acetylcholinesterase, a serine esterase found in the synapse, cleaves the transmitter, at rates close to diffusion controlled. Diisopropylphosphofluoridate (DIPF) also inhibits this enzyme, which effectively makes it a potent chemical warfare agent. Another fluoride-based inhibitor of this enzyme, sarin (Figure \(\PageIndex{26}\)), is the most potent lethal chemical agent of this class known. Only 1 mg is necessary to kill a human being.

Serine proteases have unique specificities to allow cleavage after a different subset of side chains. They cleave the peptide bond on the carboxylic acid side of specific amino acids and the specificity is determined by the size/shape/charge of amino acid side chain that fits into the enzyme’s S1 binding pocket (Figure \(\PageIndex{27}\)). Three chymotrypsin-like family members that share high sequence homology are the pancreatic digestive enzymes, trypsin, chymotrypsin and elastase. The protein cleavage sites of these enzymes vary. Trypsin cleaves proteins on the carboxylic side of basic residues, such as lysine and arginine, while chymotrypsin cleaves after aromatic hydrophobic amino acids, such as phenylalanine, tyrosine, and tryptophan. Elastase cleaves after small, hydrophobic residues, such as glycine, alanine, and valine. As shown in Figure \(\PageIndex{27}\), variations in the amino acid residues within the binding pocket of these proteases, enables electrostatic interactions with the substrate and determines sequence specificity.
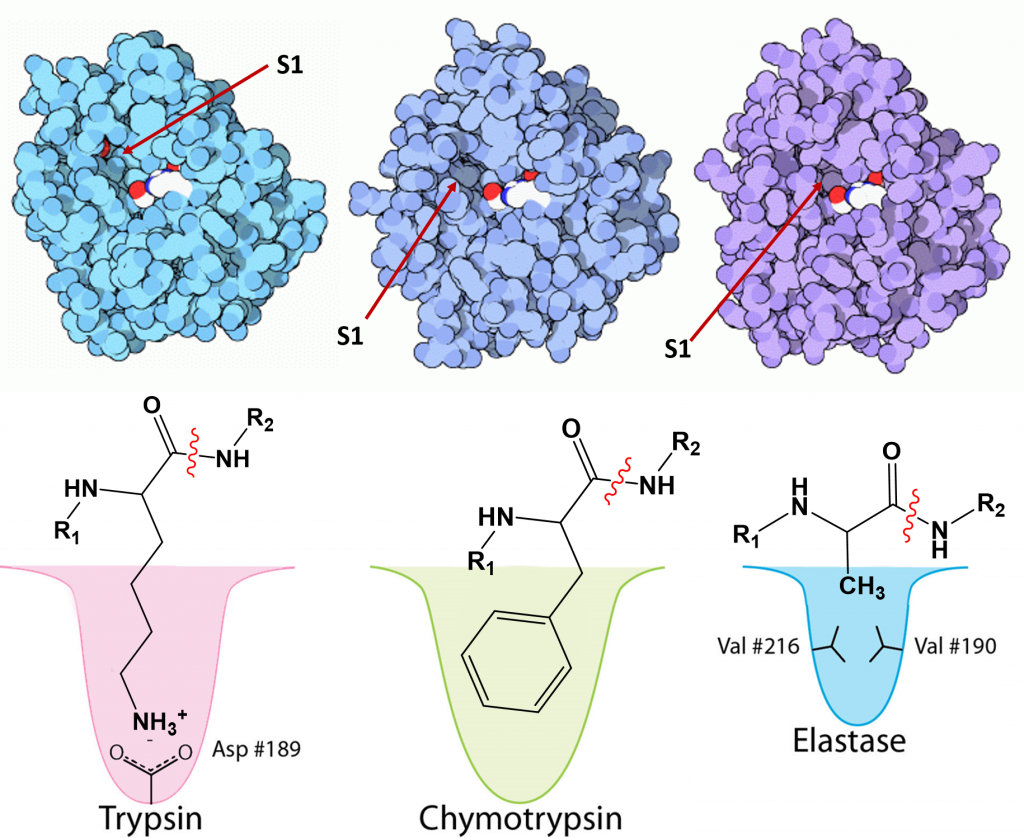
A schematic nomenclature developed by Berger and Schechter is often used to show the sites on the substrate (labeled P3, P2, P1, P1', P2' and P3') referring to the products made after cleavage of the peptide/protein that is cleaved between P1 and P' (the scissile bond) and the corresponding sites on the protease (S3, S2, S1, S1', S2' and S3'). This is illustrated in Figure \(\PageIndex{28}\).

Serine proteases are just one type of endoproteases. However, they are extremely abundant in both prokaryotes and eukaryotes. Protease A, a chymotrypin-like protease from Stremptomyces griseus, has a very different primary sequence than chymotrypsin, but its overall tertiary structure is quite similar to chymotrypsin. The positions of the catalytic triad amino acids in the primary sequences of the protein are very similar, indicating that the genes for the proteins diverged from a common precursor gene. In contrast, subtilisin, a serine protease from B. Subtilis, has both limited sequence and tertiary structure homology to chymotrypsin. However, when folded it also has a catalytic triad (Ser 221 - His 64 - Asp 32) similar to that of chymotrypsin (Ser 195 - His 57 - Asp 102). The alignment of the core structures of chymotrypsin (5cha, magenta) and subtilisin (1sbc, cyan), are shown in Figure \(\PageIndex{29}\).

The list of serine proteases is quite long. They are grouped in two broad categories - 1) those that are chymotrypsin-like and 2) those that are subtilisin-like. Though subtilisin-type and chymotrypsin-like enzymes use the same mechanism of action, including the catalytic triad, the enzymes are otherwise not related to each other by sequence and appear to have evolved independently. They are, thus, an example of convergent evolution - a process where evolution of different forms converge on a structure to provide a common function.
Proteases have multiple functions, other than in digestion, including degrading old or misfolded proteins and activating precursor proteins (such as clotting proteases and proteases involved in programmed cell death). In general, four different classes of proteases have been found, based on residues found in their active sites. Proteases can also be integral membrane proteins, and carry out their activities in the hydrophobic environment of the membrane. For example, aberrant cleavage of the amyloid precursor protein by the membrane protease presenillin can lead to the development of Alzheimer's Disease.
Table \(\PageIndex{4}\) below shows a classification of proteases based on their active site nucleophiles.
| Class (active site) | Active Site Nucleophile | Location | Examples |
| Serine/Threonine Hydrolases | Ser/Thr | soluble | trypsin, chymotrypsin, subtilisin, elastase, clotting enzymes, proteasome |
| membrane | Rhomboid family | ||
| Aspartic Hydrolases | H2O activated by 2 Asps | soluble | pepsin, cathepsin, renin, HIV protease |
| membrane | β-secretase (BACE), presenilin I, signal peptide peptidase | ||
| Cysteinyl Hydrolases | Cys | soluble | bromelain, papain, cathespsins, caspases |
| membrane | ? | ||
| Metallo Hydrolases | H2O activated by 1 or 2 metal ions | soluble | thermolysin, angiotensin converting enzyme |
| membrane | S2P family |
| Glutamate Hydrolases | Glu | . | eqolysins (fungal) |
| Asparagine Lysases (EC4) (elimination rx which are self-cleavage and hence not catalytic) | Asn | . | Tsh autotransporter E. Coli |
Table \(\PageIndex{4}\): Protease classification
How do integral membrane protease catalyze the hydrolysis (using water) of transmembrane domains in proteins, given the hydrophobic environment of the bilayer? The rhomboid class of membrane proteases, which are found in prokaryotic and eukaryotic cells, is one of the most conserved membrane proteins in nature. Instead of using a catalytic triad, these serine proteases use a dyad of Ser 201 as a nucleophile and His 254 as a general acid/base.
The chief requirement for protein substrates of rhomboids is the presence of a transmembrane domain in the target protein. No specific amino acid sequence seems to be required for specificity of one particular substrate, the drosophila transmembrane protein spitz found in Golgi membranes. On cleavage of this protein, the remaining part of the protein is released as a water soluble protein to the lumen of the Golgi where it can eventually be released from the cell. The soluble protein fragment that is released from the cell contains an epidermal growth factor domain.
The structure of a rhomboid protease, GlpG (EC:3.4.21.105), from E. Coli, was determined. It is a serine protease with a catalytic dyad (Ser 201 and His 254) instead of a triad as in most serine proteases. This transmembrane protein has 6 transmembrane helices. The enzyme has a polar active site at the bottom of a V-shape opening situated laterally in the membrane. The active site His and Ser residues are deep in this V-shaped cleft, well below the surface of the membrane. Access to the transmembrane strand of the protein substrate is blocked by a loop, which must be gated open to allow substrate access between the V-shaped gap between helices S1 and S3. Ser 201 (nucleophile) and His 254 (general base/acid) are essential for activity. The active site His 254 can be covalently modified with different chloromethylketone peptide derivatives. Figure \(\PageIndex{30}\) shows an interactive iCn3D model of the Rhomboid intramembrane protease GlpG 4QO2.
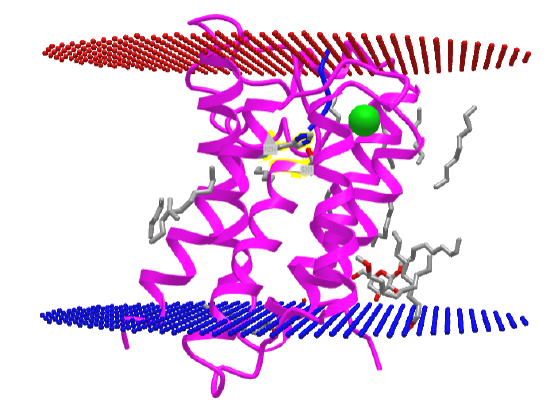
Figure \(\PageIndex{30}\): Rhomboid intramembrane protease GlpG (4QO2). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...ft9vFn7WqcujM7
Proteolytic enzymes (also termed peptidases, proteases and proteinases) are found in all living organisms, from viruses to animals and humans. Proteolytic enzymes have great medical and pharmaceutical importance due to their key role in biological processes and in the life-cycle of many pathogens. Proteases are extensively applied enzymes in several sectors of industry and biotechnology, furthermore, numerous research applications require their use, including production of Klenow fragments, peptide synthesis, digestion of unwanted proteins during nucleic acid purification, cell culturing and tissue dissociation, preparation of recombinant antibody fragments for research, diagnostics and therapy, and the exploration of the structure-function relationships.
Proteolytic enzymes belong to the hydrolase class of enzymes and are grouped into the subclass of the peptide hydrolases or peptidases. Depending on the site of enzyme action, the proteases can also be subdivided into exopeptidases (like chymotrypsin) or endopeptidases (like carboxypeptidase A) as we will discuss next. Exopeptidases, such as aminopeptidases and carboxypeptidases catalyze the hydrolysis of the peptide bonds near the N- or C-terminal ends of the substrate, respectively. Endopeptidases cleave peptide bonds at internal locations within the peptide sequence. These differences are illustrated in Figure \(\PageIndex{31}\). Proteases may also be nonspecific and cleave all peptide bonds equally or they may be highly sequence specific and only cleave peptides after certain residues or within specific localized sequences.

The action of proteolytic enzymes is essential in many physiological processes. For example, proteases function in the digestion of food proteins, protein turnover, cell division, the blood-clotting cascade, signal transduction, processing of polypeptide hormones, apoptosis and the life-cycle of several disease-causing organisms including the replication of retroviruses such as the human immunodeficiency virus (HIV). Due to their key role in the life-cycle of many hosts and pathogens they have great medical, pharmaceutical, and academic importance.
It was estimated previously that about 2% of the human genes encode proteolytic enzymes and due to their necessity in many biological processes, proteases have become important therapeutic targets. They are intensively studied to explore their structure-function relationships, to investigate their interactions with the substrates and inhibitors, to develop therapeutic agents for antiviral therapies or to improve their thermostability, efficiency and to change their specificity by protein engineering for industrial or therapeutic purposes.
The following section material (between the two horizontal lines) is not found in most biochemistry textbooks, so it could be considered optional. At the same time, it offers another unique way of understanding enzymes that ultimately will lead to a better understanding of how they function.
Enzyme catalysis in organic solvents
In our earlier lists, we mentioned changing the solvent and exploring its affect on enzyme catalysis. It might seem a bit wild, but as we saw with the rhomboid protease, some enzymes work in hydrophobic environments. Also, lipases work at the boundary between the aqueous and hydrophobic worlds. For those interested, let's see what happens when we change solvents. These including putting the enzyme in various solvents, or mixtures of solvents, as described below:
- Water miscible solvents like ethanol and acetone were added. If the water concentration was high enough, activity remained.
- Biphasic mixtures in which an aqueous solution of an enzyme was emulsified in a water immiscible solvent like chloroform or ethylacetate. The substrate would partition into both phases, while the product hopefully would end up into the organic phase.
- Nearly nonaqueous solvents, with a few % water at less than the solubility limits of water.
- Anhydrous organic solvents (0.01% water). It is this case that is most astonishing since enzymatic activity is often retained.
It is important to realize that in this last case, the enzyme is not in solution. It is rather in suspension and acts as a heterogeneous catalyst, much like palladium acts as a heterogeneous catalyst in the hydrogenation of alkenes. The suspension must be mixed vigorously and then sonicated to produce small suspended particles, so diffusion of reactants into the enzyme and out is not rate limiting. Let's explore the activity of chymotrypsin in a nonpolar solvent.
Why aren't the enzymes inactive? Surely it must seem ridiculous that they aren't, since as we learned earlier, proteins are not that stable. A 100 amino acid protein on average is stabilized only about 10 kcal/mol (41 kJ/mol) over the denatured state, or the equivalent of a few H bonds. Surely the hydrophobic effect, one of the dominant contributors to protein folding and stability, would not stabilize the native structure of enzymes in nonpolar organic solvents, and the protein would denature. It doesn't however! Maybe the real question should be not whether water is necessary, but rather how much water is necessary. The enzyme can't "see" more than a monolayer or so of water around it. The data suggests that the nature of the organic solvent is very important. The most hydrophobic solvents are best in terms of their ability to maintain active enzymes! Chymotrypsin retains 104 more activity in octane than pyridine (see kcat/Km below), which is more hydrophilic than octane. The more polar the solvent, the more it can strip bound water away from the protein. If you add 1.5% water to acetone, the bound water increases from 1.2 to 2.4%, and the activity of chymotrypsin increases 1000 fold.
Table \(\PageIndex{5}\) below shows chymotrypsin activity in organic solvents.
| Solvent | Structure | kcat/Km (M-1min-1) | relative ratio kcat/Km |
H2O bound to enzyme (%, w/w) |
| Octane |  |
63 | 15000x | 2.5 |
| Toluene |  |
4.4 | 1000x | 2.3 |
| Tetrahydrofuran | 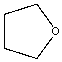 |
0.27 | 175x | 1.6 |
| Acetone | 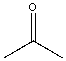 |
0.022 | 5.5x | 1.2 |
| Pyridine | 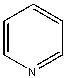 |
<0.004 | 1x (.004) | 1.0 |
Table \(\PageIndex{5}\): Chymotrypsin activity in organic solvents
Consider the following questions:
- How much water do the enzymes need? 1 molecule of chymotrypsin in octane has less than 50 molecules of water associated and can demonstrate activity. To form a monolayer requires about 500 water molecules. Water can be added which presumably leads to more bound water and higher activity.
- How stable are the enzymes? Denaturation requires conformational flexibility, which apparently requires water. The half-life of chymotrypsin in water at 60 oC is minutes, but in octane at 100 oC it is hours. At 20 oC, the half-life in water is a few days, but in octane it is greater than 6 months. Remember two factors contribute to stability: 1. The protein can denature at high temperatures. 2. Chymotrypsin is a protease, it can cleave itself in an autoproteolytic reaction.
Table \(\PageIndex{6}\) below shows the half-life of chymotrypsin activity in water and octane
| Solvent | 60oC | 100oC | 20oC |
| water | minutes | - | few days |
| octane | - | hours | > 6 months |
Table \(\PageIndex{6}\): Half-life of chymotrypsin activity in water and octane at different temperatures
- Is the enzyme specificity changed? The net binding energy is a function of the binding energy of the substrate - the binding energy of the water, since water must be displaced from the active site on binding. In an anhydrous solvent, specificity changes must be expected. For chymotrypsin, the driving force for binding of substrates in water is mostly hydrophobic. In water, the kcat/KM for the reaction of N-acetyl-L-Ser-esters is reduced 50,000 times compared to the Phe ester. However, in octane, chymotrypsin is three times more active toward Ser esters than Phe esters.
Table \(\PageIndex{7}\) shows specificity changes in chymotrypsin in water and octane
| Substrate | kcat/Km | |
| solvent: H2O | solvent: Octane | |
| N-acetyl-L-Ser-ester | 1x | 3x |
| N-acetyl-L-Phe-ester | 50,000x | 1x |
Table \(\PageIndex{7}\): Specificity changes in chymotrypsin in water and octane
Now, consider competitive inhibitors. Naphthalene binds 18 times more tightly than 1-naphthoic acid, but in octane, chymotrypsin binds naphthoic acid 310 times as tightly. Likewise the ratio of [kcat/Km (L isomer)]/[kcat/Km (D isomer)] of N-acetyl-D- or N-acetyl-L-Ala-chloroethyl esters is 1000-10,000 in water, but less than 10 in octane.
Table \(\PageIndex{8}\) shows chymotrypsin inhibition constants in water and octane.
| Inhibitor | Inhibition Constant Ki (nM) | |
| In water | In Octane | |
| Benzene | 21 | 1000 |
| Benzoic acid | 140 | 40 |
| Toluene | 12 | 1200 |
| Phenylacetic acid | 160 | 25 |
| Naphthalene | 0.4 | 1100 |
| 1-Naphthoic acid | 7.2 | 3 |
Table \(\PageIndex{8}\): Chymotrypsin inhibition constants in water and octane
Can new reactions be carried out in nonpolar solvents? The quick answers is yes, since reactions in aqueous solutions can be unfavorable due to low Keq values, side reactions, or insolubility of reactants. Consider lipases, which cleave fatty acid esters by hydrolysis in aqueous solutions. In nonaqueous solutions, reactions such as transesterification or ammonolysis can be performed.
Enzymes are clearly active in organic solvents which appears to contradict our central concepts of protein stability. Two reasons could could explain this stability:
- It is possible that from a thermodynamic view, the enzyme is stable in organic solvents. However, as was discussed above, this is inconceivable given the delicate balance of noncovalent and hydrophobic interactions required for protein stability.
- The second reason must win the day: the protein is unable to unfold from a kinetic point of view. Conformational flexibility is required for denaturation. This must require water as the solvent. Denaturation in organic solvents is kinetically, not thermodynamically controlled.
A specific example helps illustrate the effects of different solvents on chymotrypsin activity. Dry chymotrypsin can be dissolved in DMSO, a water miscible solvent. In this solvent it is completely and irreversibly denatured. If it is now diluted 50X with acetone with 3% water, no activity is observed. (In the final dilution, the concentrations of solvents are 98% acetone, 2.9% water, and 2% DMSO.) However, if dry chymotrypsin was added to a mixture of 98% acetone, 2.9% water, and 2% DMSO, the enzyme is very active. We end up with the same final solvent state, but in the first case the enzyme has no activity while in the second case it retains activity. These ideas are illustrated in Figure \(\PageIndex{32}\).

Dry enzymes added to a concentrated water-miscible organic solvent (like DMSO) will dissolve and surely denature, but will retain activity when added to a concentrated water-immiscible solvent (like octane), in which the enzyme will not dissolve but stay in suspension.
It appears the enzymes have very restricted conformational mobility in nonpolar solvents. By lyophilizing (freeze-drying) the enzyme against a specific ligand, a given conformation of a protein can be trapped or literally imprinted onto the enzyme. For example, if the enzyme is dialyzed against a competitive inhibitor (which can be extracted by the organic solvent), freeze-dried to remove water, and then added to a nonpolar solvent, the enzyme activity of the "imprinted" enzyme in nonpolar solvents is as much as 100x as great as when no inhibitor was present during the dialysis. If chymotrypsin is lyophilized from solutions of different pHs, the resulting curve of V/Km for ester hydrolysis in octane is bell-shaped with the initial rise in activity reaching half-maximum activity at a pH of around 6.0 and a fall in activity reaching half-maximum at pH of approximately 9.
Use of enzymes in organic solvent allows new routes to organic synthesis. Enzymes, which are so useful in synthetic reactions, are:
- stereoselective - can differentiate between enantiomers and between prochiral substrates
- regioselective - can differentiate between identical functional groups in a single substrate
- chemoselective - can differentiate between different functional groups in a substrate (such as between a hydroxyl group and an amine for an acylation reaction)
Enzyme in anhydrous organic solvents are useful (from a synthetic point) not only since new types of reactions can be catalyzed (such as transesterification, ammonolysis, thiolysis) but also because the stereoselectivity, regioselectivity, and chemoselectivity of the enzyme often changes from activities of the enzyme in water.
Organic reactions are usually conducted in organic solvents, since many organic molecules react with water, and the reagents and products are usually not soluble in water. In a manner analogous to using an enzyme as a heterogeneous catalyst in nonpolar solvent, Sharpless is pioneering a technique to conduct organic reactions in water. They (Narayan et al.) have shown that many unimolecular and bimolecular reactions occur faster in water than in organic solvents. As in enzyme catalysis in nonpolar solvent, the reactions must be mixed vigorously to disperse reactants in micro-drops (a suspension) in water, greatly increasing the surface area that might allow water to act on transition states or intermediates to stabilize them through hydrogen bonding. They called these reactions "on water" reactions since reactants usually float on water. They have performed cycloadditions, alkene reactions, Claisen rearrangments, and nucleophilic substitution reactions using this process. One cycloaddition reaction went to completion in ten minutes at room temperature, compared to 18 hours in methanol and 120 in toluene. Adding nonpolar solvent at certain times greatly increased the rate of the reaction.
Carboxypeptidase A
This enzyme (EC 3.4.17.1) cleaves the C-terminal amino acid from a protein through a hydrolysis reaction. As such it is an exoprotease (not an endoprotease which cleaves proteins internally within the sequence). In terms of selectivity toward C-terminal amino acids, its activity is increased if the C-terminal side chain group is aromatic or branched aliphatic (Phe, Tyr, Trp, Leu or Ile). X-Ray structures of the enzyme with and without a competitive inhibitor show a large conformational change at the active site when inhibitor or substrate is bound. Without inhibitor, several waters occupy the active site. When an inhibitor (and presumably, by extension, a substrate) is bound, the water leaves (which is entropically favored), and Tyr 248 swings around from near the surface of the protein into the active site to interact with the carboxyl group of the bound molecule, a distance of motion equal to about 1/4 the diameter of the protein. This effectively closes off the active site and expels the water.
A Zn2+ ion is present at the active site. It is bound by His 69, His 196, Glu 72, and finally a water molecule as the fourth ligand. A hydrophobic pocket that interacts with the phenolic group of the substrate accounts for the specificity of the protein. In the catalytic mechanism, Zn2+ might have several roles. In one, it may help a coordinated water to be more nucleophilic by either polarizing the water or converting it to a more potent nucleophile OH-. It might also stabilize developing negative charges in the transition state and in an intermediate. Two possible mechanisms have been offered.
The Water Pathway. In this proposed mechanism, water acts as a nucleophile, and is deprotonated by Glu 270, acting as a general base. Glu 270, along with Zn2+, helps to promote dissociation of a proton from the bound water, making it to a better nucleophile. Water attacks the electrophilic carbon of the sessile bond, forming a tetrahedral intermediate. The tetrahedral intermediate then collapses, expelling the alkoxy leaving group, which picks up a proton from Glu 270, now acting as a general acid catalyst. People used to believe that Tyr 248 acted as a general acid, but mutagenesis showed that Tyr 248 can be replaced with Phe 248 without significant effect on the rate of the reaction. A simplified reaction reaction is shown in Figure \(\PageIndex{33}\).

Figure \(\PageIndex{33}\): Water pathway mechanism for carboxypeptide A. After Wu et al. J Phys Chem B. 2010 July 22; 114(28): 9259–9267. doi:10.1021/jp101448j
Nucleophilic Pathway. In this pathway, Glu 270 is the primary initial nucleophile in the formation of the initial tetrahedral intermediate. The role of Zn2+ is in charge stabilization. This pathway is illustrated in Figure \(\PageIndex{34}\).

Figure \(\PageIndex{35}\) shows an interactive iCn3D model of the active site of bovine carboxypeptidase in the absence of a substrate or inhibitor (1M4L). The Zn2+ ion is shown as a red sphere.
.png?revision=1&size=bestfit&width=335)
Figure \(\PageIndex{35}\): Bovine carboxypeptidase A (1M4L) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...sX1NYbGav1CFY6
Note how far Tyr248 is away from the active site in the model. Glu72 and Glu270 are negatively charged in the resting state of the enzyme at pH 7.5. The values are much higher (weaker acid) than solution pKa of the side chain of glutamic acid. Also the water bound to the Zn2+ is long enough to suggest that the water is neutral and not in the form of OH- in this form of the enzyme. If OH- were present, the distance between it and the Zn2+ would be shorter due to the great electrostatic force.
Figure \(\PageIndex{36}\) shows an interactive iCn3D model of the active site of bovine carboxypeptidase bound to the inhibitor aminocarbonylphenylalanine (1HDU). The Zn2+ ion is shown as a red sphere.
.png?revision=1&size=bestfit&height=320)
Figure \(\PageIndex{36}\): Bovine carboxypeptidase A bound to bound to the inhibitor aminocarbonylphenylalanine (1HDU). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...vWt2qeZtUrwj78
Note the closer proximity of tyrosine 248 to the active site.
Lysozyme
Lysozyme (EC 3.2.1.17), found in cells and secretions of vertebrates but also in viruses which infect bacteria, cleaves peptidoglycan GlcNAc (β-1,4) MurNAc repeat linkages (NAG-NAM) in the cell walls of bacteria and the GlcNAc(β-1,4) GlcNAc (poly-NAG) in chitin, found in the cells walls of certain fungi. Since these polymers are hydrophilic, the active site of the enzyme would be expected to contain a solvent-accessible channel into which the polymer could bind. The crystal structures of lysozyme and complexes of lysozyme and NAG have been solved to high resolution. The inhibitors and substrates form strong H bonds and some hydrophobic interactions with the enzyme cleft. Kinetic studies using (NAG)n polymers show a sharp increase in kcat as n increases from 4 to 5. The kcat for (NAG)6 and (NAG-NAM)3 are similar. Models studies have shown that for catalysis to occur, (NAG-NAM)3 binds to the active site with each sugar in the chair conformation, except the fourth which is distorted to a half chair form. This labilizes the glycosidic link between the 4th and 5th sugars. Additional studies show that if the sugars that fit into the binding site are labeled A-F, then because of the bulky lactyl substituent on the NAM, residues C and E cannot be NAM, which suggests that B, D and F must be NAM residues. Cleavage occurs between residues D and E.
A review of the chemistry of glycosidic bond (an acetal) formation and cleavage shows the acetal cleavage is catalyzed by acids and proceeds by way of an oxonium ion which exists in resonance form as a carbocation. A reaction mechanism of hemiacetal/acetal formation and cleavage is illustrated in Figure \(\PageIndex{37}\).

Catalysis by the enzyme involves Glu 35 and Asp 52 which are in the active site. Asp 52 is surrounded by polar groups but Glu 35 is in a hydrophobic environment. This should increase the apparent pKa of Glu 35, making it less likely to donate a proton and acquire a negative charge at low pH values, making it a better general acid at higher pH values. Here is a possible general mechanism:
- binding of a hexasaccharide unit of the peptidoglycan with concomitant distortion of the NAM.
- protonation of the sessile acetal O by the general acid Glu 35 (with the elevated pKa), which facilitates cleavage of the glycosidic link and formation of the resonance stabilized oxonium ion.
- Asp 52 stabilizes the positive oxonium through electrostatic catalysis. The distorted half-chair form of the NAM stabilizes the oxonium which requires co-planarity of the substituents attached to the sp2 hybridized carbon of the carbocation resonant form (much like we saw with the planar peptide bond).
- water attacks the stabilized carbocation, forming the hemiacetal with release of the extra proton from water to the deprotonated Glu 35 reforming the general acid catalysis.
Part of a mechanism illustrating the roles of Glu 35 and Asp 52 is shown below in Figure \(\PageIndex{38}\).

Binding and distortion of the D substituent of the substrate (to the half chair form as shown above) occurs before catalysis. Since this distortion helps stabilize the oxonium ion intermediate, it presumably stabilizes the transition state as well. Hence this enzyme appears to bind the transition state more tightly than the free, undistorted substrate, which is yet another method of catalysis.
pH studies show that side chains with pKa's of 3.5 and 6.3 are required for activity. These presumably correspond to Asp 52 and Glu 35, respectively. If the carboxy groups of lysozyme are chemically modified in the presence of a competitive inhibitor of the enzyme, the only protected carboxy groups are Asp 52 and Glu 35.
In an alternative mechanism, Asp 52 acts as a nucleophilic catalyst and forms a covalent bond with NAM, expelling a NAG leaving group with Glu 35 acting as a general acid as shown in Figure \(\PageIndex{39}\). This alternative mechanism also is consistent with other β-glycosidic bond cleavage enzyme. Substrate distortion is also important in this alternative mechanism.

Figure \(\PageIndex{39}\): Alternative mechanism for lysozyme catalysis employing Asp 52 as a nucleophilc catalyst (after Vocadlo et al., Nature, 412 (2001), https://www.nature.com/articles/35090602.
Recent structural work shows that Asp 52 is involved in a strong hydrogen bond network that might preclude its ability to form a covalent bond with the glycan substrate. An earlier structure (1H6M) did show a covalent bond.
Figure \(\PageIndex{40}\) shows an interactive iCn3D model of the active site of hen egg white lysozyme bound to a (NAG)4 glycan (7BR5). Note the positions of E35 and D52.
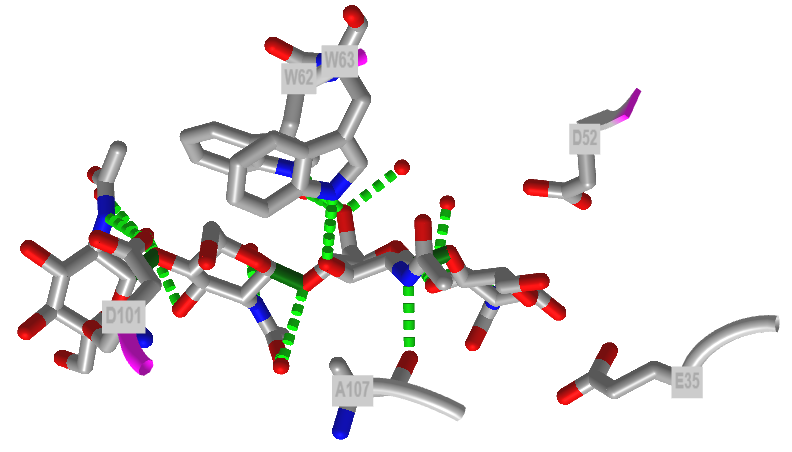
Figure \(\PageIndex{40}\): Interactions of hen egg white lysozyme with bound (NAG)4 glycan (7BR5) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?mnZuP4W4pfTxKRmX7
Summary
In this section, we explored some of the biochemical arrow pushing conventions for SN2 reactions, including methyltransferase enzymes that are dependent on the coenzyme SAM, and kinases, which transfer a phosphate group. We saw that the nucleophilic acyl substitution mechanisms for chymotrypsin and carboxypeptidase can be quite abbreviated such that it takes some biochemical intuition to recognize that a tetrahedral intermediate is operative. There are two major proposed mechanisms for carboxypeptidase, which propose distinct roles for the metal ion in the reaction.
The investigation of chymotrypsin's mechanism by substrate specificity experiments (changing the substrate), altering the pH, mutagenesis and reaction with irreversible inhibitors (changing the protein) gives rich information that can be used to deduce enzyme mechanisms. Coupled with structural data, these investigations reveal key details about enzyme structure-function, and allow biochemists to propose reasonable enzyme mechanisms.