
28.2: At the cell membrane- receptors and receptor enzymes
- Page ID
- 14991


Introduction
We are going to start a more detailed description of cell signaling where it begins, at the cell membrane, and move inward, toward intracellular organs, where we will end at the nucleus and changes in gene expression mediated by the signal. Of course, this end is somewhat arbitrary as the signal could propagate from the nucleus back to the membrane where newly synthesized membrane protein might be inserted or even exported, as in the case of secreted antibodies. It is difficult enough to keep track of all the molecular players, let alone add onto that their initial location in the cell and their final location if they translocate. Signaling can also be more daunting for those with a more chemistry focus, who find the details of cellular structure and trafficking a bit daunting. Figure \(\PageIndex{1}\) shows a truncated view of the cell membrane, membrane proteins, and some of the organelles that we will visit throughout this chapter. To stay localized we will repeat the figure or variants of it several times in this section.

Receptors at Cell Membrane
Let's start at the location where signaling almost invariable begins, in the cell membrane, where signals (hormones, neurotransmitters, nutrients, other cells) bind to cell surface transmembrane proteins shown in the red box in Figure \(\PageIndex{2}\).

We have already discussed integral and peripheral membrane proteins in Chapter 11. When occupied by a ligand signal, the signaling event mediated by a transmembrane receptor might be a change in the membrane protein conformation, which propagates to its intracellular domain. Alternatively, the receptor might change its conformation and become a ligand-gated kinase (or possibly a phosphatase) or a ligand-gated channel or pore. We will discuss signal-gated ion channels in Chapter 28.9 - Neural Signaling.
Ultimately, intracellular enzymes are activated in cells in response to the external molecular signal. This provides amplification of the initial signal since a single activated enzyme undergoes multiple rounds of catalysis before it would become inactivated. If the multiple products (a second messenger or phosphorylated proteins for example) that are formed activate multiple addition enzymes, the signal is further amplified.
Receptors with no kinase or transport activity - G Protein-Coupled Receptors (GPCRs)
One major type of signaling receptor is the G-protein coupled receptor (GPCR). Over 800 GPCRs are encoded in the human genome which represents almost 4-5% of the number of protein-coding genes. The proteins below to five major families: rhodopsin, secretin, glutamate, adhesion, and frizzled/taste2. They don’t express enzymatic activity but on binding, they can activate enzymes inside the cell by interacting through their cytoplasmic domains with G proteins (GTP binding proteins) in the cytoplasm. GPCRs have been called serpentine receptors as their single amino acid chain proteins have 7 transmembrane-spanning α- helices. All of the GPCRs have similar yet slightly different structures, allowing them to interact with specific ligands. Many of the GPCRs bind to unknown ligands and hence are called orphan receptors. We will explore a few GPCRs in more detail below.
GPCRs that modulate the membrane enzyme adenylyl cyclase:
β-adrenergic receptor
The β-adrenergic receptor is a prototypical GPCR. Found in muscle, liver, and fat cells, it binds epinephrine and adrenaline, which leads to energy mobilization and muscle activation (i.e. flight or fight response). The mechanism of activation of a GPCR is illustrated using the beta-adrenergic receptor as an example. The unoccupied adrenergic receptor is associated with a heterotrimeric G protein, which contains an α, β, and γ subunits. GDP is usually bound to the α subunit. Figure \(\PageIndex{3}\) shows a cartoon of the GPCR interacting with the heterotrimeric G protein.

When the hormone is bound to the receptor, a conformation change is propagated to the cytoplasmic domain of the GPCR altering its interaction with Gαβγ. This causes a conformational change in the Gα subunit, which leads to an exchange of bound GDP with GTP in the Gα subunit, promoting the dissociation of the Gα-GTP from Gβγ. Gα-GTP then binds to and activates an adjacent membrane enzyme called adenylate cyclase, which produces a second messenger by converting ATP to cyclic AMP (cAMP). Figure \(\PageIndex{4}\) shows steps in the generic GPCR activation cycle.
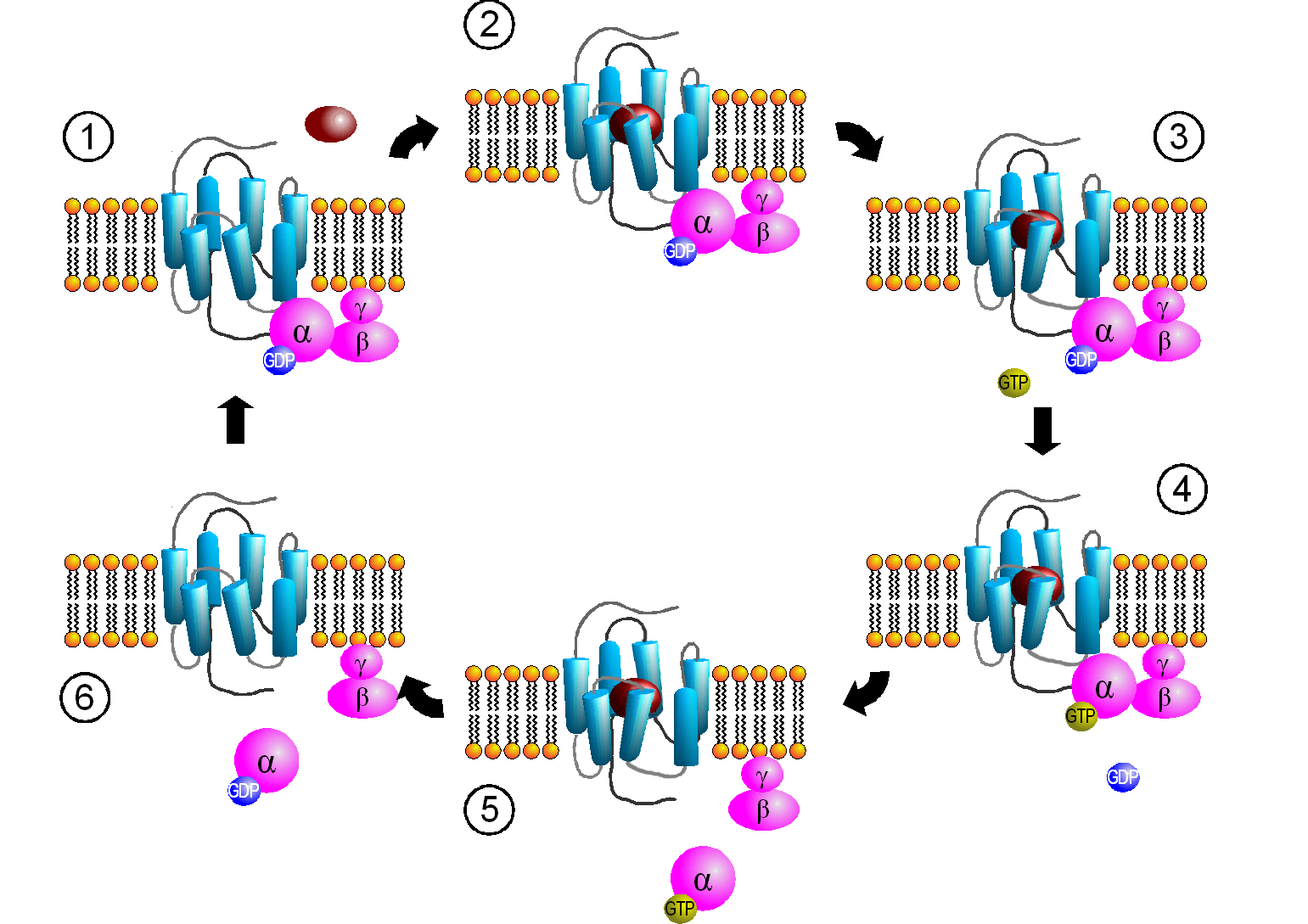
Figure \(\PageIndex{4}\): Activation cycle of G-proteins by G-protein-coupled receptors. https://commons.wikimedia.org/wiki/F...PCR-Zyklus.png Creative Commons Attribution-Share Alike 3.0 Unported
The primary message binds to the GPCR (1 leading to state 2. The cytoplasmic domain conformational changes allow GTP exchange in the Gα subunit as state 3 goes to 4. In 5 the Gα-GTP complex dissociates from Gβγ, which remains in the membrane. The Gα-GTP subunit is held and localized to the membrane (not evident in the above figure) through a lipid anchor attached through a post-translational modification.
Remember, the GPCR has no ligand-gated enzymatic activity. Yet it indirectly leads to the activation of a membrane enzyme, adenylate cyclase, when the dissociated Gα-GTP binds to the cyclase. As long as GTP remains bound to the Gα subunit, it will continue to modulate the activity of adenylate cyclase. A built-in regulatory mechanism does exist in the protein since the Gα subunit has GTPase activity. The GTP will eventually hydrolyze, and the GDP-Gα subunit will lose affinity for its bound partner (adenylate cyclase), and return to the heterotrimeric G protein associated with the unbound receptor. GPCRs bind the signaling ligand (primary message) in a binding cavity localized at the extracellular face and between four of the transmembrane helices.
The activity and structure of GPCRs have been studied using natural ligands (hormones and neurotransmitters), as well as agonists, partial agonists, inverse agonists, and antagonists. As discussed previously, agonists bind to the natural ligand binding site and elicit the same or a partial response (partial agonist). Inverse agonists bind and lower the response of a constitutively active receptor, and antagonists bind and prevent the normal response of an agonist. About 35% of pharmaceutical drugs target GPCRs, but target only about 15% of the ∼800 human GPCRs. The orphan GPCRs are increasingly targets for drug development. Most hormones and neurotransmitters work through GPCRs. In addition, our primary senses of vision, smell (olfaction), and taste (gustation) work through GPCRs.
Figure \(\PageIndex{5}\) shows the structure of the beta 2-andrenergic:Gs complex with bound agonist. No membrane is shown.
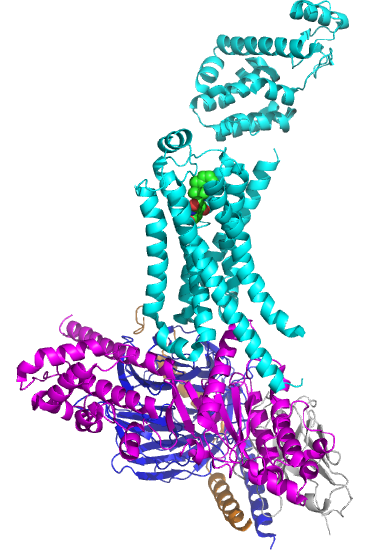
The GPCR is shown in cyan. The 7 transmembrane helices should be obvious. The ligand is bound between them. The Gα subunit is shown in magenta, the Gβ in dark blue, and the Gγ in orange/brown. The gray subunit is a Camelid antibody VHH fragment (a single-chain nanoantibody used to stabilize the conformation for crystallization. )
The biggest conformational change that occurs when the GPCR binds an agonist is an outward movement (14 Å) at the intracellular domain of transmembrane segment 6 (TM6) and an extension of the TM5 helix. This leads to a movement of the Gα's alpha-helical domain, enabling GTP exchange for GTP. Of course, multiple reactions determine the fraction of the Gα in the active GTP-bound state. These would include the relative cellular concentrations of free cytoplasmic GDP and GTP, their KD values for the Gα, the rate constant for the hydrolysis of bound GTP, and the rate constants for the GDP ↔ GTP exchange.
Figure \(\PageIndex{6}\) shows an interactive iCn3D model of the Crystal structure of the beta2 adrenergic receptor-Gs protein complex 3SN6

The GPCR is shown in green, the Gα in magenta, Gβ in blue, and the Gγ in brown. An agonist (spacefill) is shown in CPK colors near the outer leaflet.
Some bacterial toxins work by inactivating the GTPase activity of the Gα subunit, keeping it in the "stuck" position. For example, cholera toxin, an enzyme released by Vibrio cholera, catalyzes the ADP ribosylation of an Arg in the Gα subunit by transferring everything but the nicotinamide from NAD+ to the Arg residue.
Since the Gα subunit stimulated the activity of adenylyl cyclases, it is often named a stimulatory Gα protein or Gsα. Figure \(\PageIndex{7}\) shows an interactive iCn3D model of the adenylyl cyclase activator Gsα with GTP-γ-S (1azt)
.png?revision=1)
Now we can explore how the occupied GPCR, which again has no enzymatic activity, activates the enzyme adenylyl cyclase. Figure \(\PageIndex{8}\) shows a cartoon of the Gsα subunit bound to adenylyl cyclase as the Gβγ heterodimer remains associated with the membrane

Adenylate cyclase converts ATP into the second messenger cyclic AMP (cAMP) as shown in Figure \(\PageIndex{9}\). The figure also shows how cAMP is broken down into AMP by the enzyme cAMP-specific 3',5'-cyclic phosphodiesterase (PDE). The latter step is necessary to control the lifetime of the second messenger cAMP.

You might ask why cAMP and not just AMP is nature's choice for GPCR signaling. AMP is a very important metabolic species. High concentrations of it signal an energy-depleted state. AMP is used in another signaling process to mobilize a response to adjust the energy state of a cell using a protein called AMP-Protein Kinase, which we will describe later. Many enzymes are also allosterically regulated by AMP.
Figure \(\PageIndex{10}\) shows an interactive iCn3D model of the membrane adenylyl cyclase bound to an activated stimulatory Gsα protein 6R3Q determined by cryo-EM.
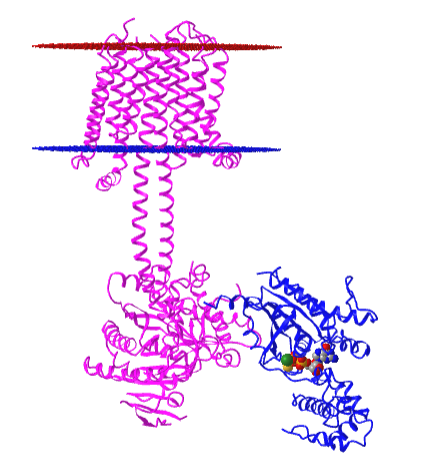
The carboxyl-terminal cytoplasmic domain has the catalytic and allosteric sites.
Cannabinoid Receptors
In contrast to the beta-adrenergic receptor which mediates activation of adenylate cyclase through Gsα, some Gα subunits inhibit adenylate cyclase when bound. These Gα subunits are called Gi/oα in contrast to the stimulatory subunits, Gsα. Also, Gα subunits interact with many proteins other than adenylate cyclase. Examples include cannabinoid receptors.
Cannabinoid receptors are named after the exogenous and psychoactive drug Δ9-tetrahydrocannabinol (THC) that binds to the receptor. THC is the major phytocannabinoid (from plants) found in the Cannabis sativa plant and the marijuana-derived from it. The other main cannabinoid in the plant is cannabidiol (CBD). Phytocannabinoids bind to two types of human cannabinoid (CB) receptors, CB1 and CB2. They have 44% amino acid and 68% homologies in the entire protein and the transmembrane domain, respectively.
The phytocannabinoids exert their effect through binding to human CB1 and CB2, whose endogenous ligands are two fatty acids derivatives called anandamide (AEA) and 2-arachidonoyl glycerol (2-AG). The structure of THC, CBD, and the two major endogenous ligands are shown in Figure \(\PageIndex{11}\).

Figure \(\PageIndex{11}\): Structure of agonist and antagonist for cannabinoid receptors
The cannabinoids from Cannabis sativa have a monoterpene isoprenyl group (C10) and a pentyl side chain (C5). The ligands are largely hydrophobic and probably access their binding site in the receptor mainly by lateral movement in the membrane. The receptors differ most in the N-terminal extracellular loop which is also involved in ligand binding.
Figure \(\PageIndex{12}\) shows an interactive iCn3D model of the class A GPCR Cannabinoid Receptor-Gi Complex Structure with bound agonist (6KPF)
.png?revision=1&size=bestfit&width=370)
The gray protein is a nanobody used to stabilize the protein during crystallization. The agonist is AM12033, which is similar to AM11542 in the figure above, but with a -C=N terminus instead of a bromide.
Cannabis sativa contains the psychoactive drug, Δ9-tetrahydrocannabinol (THC), which is a partial agonist for CB1 (binds with reported Ki values of 10 or 53 nm)and CB2 (Ki = 40 nm). Its psychoactive effects on mental activity as well as pain and appetite are well known. In contrast, cannabidiol (CBD) is the main, non-psychoactive cannabinoid. It has a much lower affinity for the recombinant CB1 (Ki = 1.5 µM ) and CB2 (Ki = 3.7 µM). It appears to be a partial antagonist for CB1 and a weak inverse agonist for CB2. It has also been shown that CBD is a negative allosteric modulator of the agonistic effects of THC and 2AG. The actual psychotropic effects of combining THC and CBD are complicated and not understood well.
Figure \(\PageIndex{13}\) shows an interactive iCn3D model of human CB1 in complex with agonist AM11542 (5XRA)
.png?revision=1&size=bestfit&width=397&height=394)
Figure \(\PageIndex{13}\): Human CB1 in complex with agonist AM11542 (5XRA). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...wDv9fXNPaMhwm9
The complex shows a significant conformational change in the structure compared to the receptor bound to an antagonist. This includes a large reduction (50%) in the volume of the binding pocket and an increase in the surface area of the receptor that would bind to G proteins.
How much of a receptor is bound with a cannabinoid depends on the concentration of the cannabinoid ligand and the Ki for the drug. The amount of THC and CBD depends on the genetics of the plant, which has been engineered to greatly decrease THC production (in the hemp plant used for nonpharmacological commercial properties) or increase either THC or CBD production at the expense of the other.
The synthesis of THC and CBD proceeds through a common precursor, CBGA (cannabigerol acid). Two key flavoproteins, Δ9-tetrahydrocannabinolic acid synthase (THCAS) and cannabidiolic acid synthase (CBDAS) convert this common precursor CBGA into two new precursors, Δ9-THCA and CBDA, respectively. This final synthetic step involves an oxidative cyclization reaction using O2 and produces H2O2. Spontaneous, non-catalyzed decarboxylation and rearrangements of Δ9-THCA and CBDA lead to the final products, THC and CBD. This last process occurs on exposure to heat, which occurs in smoking and baking, and at a slower rate during storage.
Commercially used preparations of THC and CBD for medicinal purposes vary widely in concentrations. THC concentration ranges for pain management (<5-10%) are much lower than those for psychotropic effects (<15%), with values of 21% or often found in "recreational" cannabis. High-potency THC strains can contain up to 25-30% THC by dry weight. For strands modified for CBD production, the maximal amount is about 25%. Even though CBD appears to be a partial antagonist for CB1, it appears that the ratio of THC:CBD is important in modulating the "high" or intoxicating state of THC. A ratio of THC:CBD of just over 1:1 leads to synergism or enhancement of the acute effects of THC whereas ratios of THC:CBD of 1:2 to 1:6 seem to have the least intoxicating effects. However, CBD decreases psychotic symptoms of THC and also decreases memory changes associated with THC. CBD also is an allosteric modulator of the μ-opioid receptor.
At present, there are no structures of CBD bound to its cannabinoid receptors. Figure \(\PageIndex{14}\) shows an interactive iCn3D model of the CBD-bound full-length rat transient receptor potential vanilloid 2 (TRPV2) in nanodiscs (6U88)
.png?revision=1&size=bestfit&width=425&height=348)
Figure \(\PageIndex{14}\): CBD-bound full-length rat TRPV2 in nanodiscs (6U88). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...BJcXBriq49LtQA
This receptor is a calcium-permeable non-selective cation channel that is activated at high temperatures (500C) but not vanilloids like capsaicin. Hence it acts as a "noxious high temperature" receptor.
Now let's make it even more complicated. There are more than 20 different Gα-like proteins known in 4 major families.
- Gs and Gi regulate adenylyl cyclase
- Gq activates phospholipase Cβ (described below). There are 4 members given these strange names: Gq, G11, G14, and G15/16
- G12/13 activate small GTPase protein (described in Chapter 28.5)
The Gα protein involved in light sensation is named transducin, while those involved in odorant detection and taste are called Golfactory and Ggustatory, respectively.
As we add more variants of each signaling component, the origin of the complexity of signaling systems becomes evident. For example, the neurotransmitter serotonin binds to its receptor, a GPCR, and instead of gating the protein open to ion flow (as with other ligand-gated ion channels in the activation of neurons as we will see in Chapter 28.9), it interacts with two different alpha subunits, Gs, which leads to activation of adenylyl cyclase, and G12, which interacts with other small GTP binding proteins called GEFs (we will also see these later).
GPCRs modulate the activity of the membrane enzyme phospholipase C.
These receptors use the same mechanism for activation of the membrane enzyme adenylyl cyclase. When the primary signal is bound to the GPCR, a conformation change is propagated to the cytoplasmic domain of the GPCR, altering its interaction with Gαβγ in which the alpha subunit is a member of the Gα(q) family. This causes a conformational change in the Gα subunit which leads to an exchange of bound GDP with GTP in the Gα subunit, promoting the dissociation of the Gα-GTP. Gα-GTP then binds to and activates an adjacent membrane enzyme, phospholipase C (PLC), which cleaves membrane phospholipids to produce two, second messengers, diacylglycerol and inositol 1,4,5-trisphosphate (IP3). Their structures are shown in Figure \(\PageIndex{15}\).

There appear to be 13 kinds of mammalian phospholipase Cs divided into six isotypes (β, γ, δ, ε, ζ, η). Phospholipase C is also named 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase.
Figure \(\PageIndex{16}\) shows an interactive iCn3D model of the Gα(q)-phospholipase C-β 3 structure (4GNK). The magenta structure is the Gα(q) protein with bound GDP in spacefill. The cyan structure is the Pleckstrin Homology (PH) domain of the protein. This domain targets proteins to inositol phospholipids in the membrane but appears not to have this function in this protein.
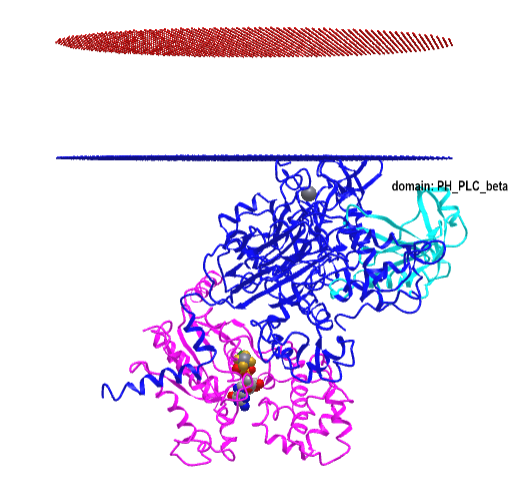-phospholipase_C-beta_3_structure_(4GNK)_with_PK_domain.png?revision=1&size=bestfit&width=405&height=367)
Note that in contrast to adenylyl cyclase, PLC is a peripheral, not an integral membrane protein. It is found in the cytoplasm as well as associated with the inner leaflet of the cell membrane where its main activities, regulating and cleaving PIP2, occur. PLC localizes to lipid rafts enriched in PIP2. Figure \(\PageIndex{17}\) shows the domain structure of phospholipase C β3

The N-terminal PH_14 represents the Pleckstrin Homology (PH) domain, which is among the top 15 of all domains in the human genome. All PLCs except PLCζ have this domain. Note that in another example of complexity, PLC-β binding to the inner leaflet does not require PIP2. The iCn3D model above indeed shows binding to the membrane seems to depend on adjacent structures and not the PH domain (cyan) specifically.
Tabel \(\PageIndex{1}\) below characteristics of common signals that signal through GPCRs.
| signal | vasopressin | epinephrine | light | odorant | odorant | sweet tastant |
| receptor | VR | β-adrenergic | rhodopsin | odorant receptor 1 | odorant receptor 2 | sweet receptor |
| Ga-like subunit | Gi | Gs | transducin | Golfactory | Golfactory | Ggustatory |
| coupled enzyme | adenylate cyclase | adenylate cyclase | phosphodiesterase | phospholipase C | adenylate cyclase | adenylate cyclase |
| 2nd messenger | decrease cAMP | increase cAMP | decrease cGMP | increase IP3 | increase cAMP | increase cAMP |
| protein affected | decrease PrK-A | increase PrK-A | dec. Ca, Na perm. | inc. Ca perm | inc.Ca, Na perm | dec. K perm |
Tabel \(\PageIndex{1}\): Characteristics of common signals that work through GPCRs.
Changeux and Edelstein reviewed the MHC model 40 years after its conception and support its application to signal transduction processes. They include in signaling molecules not only hemoglobin, but regulatory enzymes (aspartate transcarbamylase, phosphofructokinase, LDH, glycogen phosphorylase), membrane receptors (acetylcholine receptor, rhodopsin), and nuclear receptors (lac repressor, steroid hormone receptors). In all these signaling proteins, residue distant from the "active" site participates in binding to allosteric ligands. Often the allosteric site is on a separate domain that can be cleaved from the protein and still maintain allosteric ligand binding properties. The proteins also consist of multiple subunits easily related by distinct symmetry axes. Allosteric ligands often bind in cavities in subunit interfaces along symmetry axes. In general, crystal structure analyses show that low-affinity T and high-affinity R forms of the signaling proteins exist but are accompanied by minor tertiary structure changes in individual subunits (i.e. perfect symmetry in all subunits is not preserved on the binding of allosteric ligand). For neurotransmitter membrane receptors, these two states can be correlated with an open and closed state (for ion flux), and open conformations of these proteins can often be found in mutant forms. However, for many ligand-gated ion channels and G-protein coupled receptors (serpentine), kinetic analyses show more complicated forms than can be represented by a simple two-state (R and T) model. High-resolution microscopy shows evidence for nonsymmetrical quaternary structural changes. These changes can be observed in the absence of ligand, which gives support to the MWC concept that allosteric ligands select certain conformational states, leading to equilibrium shifts in the unliganded receptor to the more high-affinity state. More refined methods of structural analysis will presumably show more evidence of subtle tertiary changes in the proteins that are preludes to quaternary structural changes. Yet the simplicity of the MWC model for explaining many features of signaling proteins remains.
Receptors with signal-gated kinase activity - Receptor Tyrosine Kinases (RTK)
Why bother binding a primary message to a GPCR and going through multiple steps before the activation of a membrane enzyme like adenylate cyclase? Wouldn't it be easier and more efficient to have the membrane receptor a ligand-activated enzyme? Such is the case with special membrane receptors called Receptor Tyrosine Kinases (RTKs). There are about 90 tyrosine kinases in the human genome of which 58 are RTKs. Figure \(\PageIndex{18}\) shows the family domain structure of the RTKs.

Note that the insulin receptor (InsR) is a dimer of two monomeric insulin receptor chains. Figure \(\PageIndex{19}\)s shows in more detail the domain structure of the epidermal growth factor receptor (EGFR).

Figure \(\PageIndex{19}\): Domain structure of the EGFR domain.
The domain (red/brown) immediately after the blue domain is the transmembrane domain. Furin is a cellular endoprotease. The green represents L domains which comprise the ligand binding site. Each L domain consists of a single-stranded right-hand beta-helix.
Here is the cascade of events for signaling through EGFR: The transmembrane has ligand-dependent tyrosine kinase activity. Binding of the hormone EGF causes receptor dimerization bringing the intracellular kinase domains (yellow PK_Try_Ser_Thr) together activating them. When active, they can phosphorylate each other (autophosphorylation) or other proteins. When the receptor is autophosphorylated, other proteins can bind to the cytoplasmic domain of the receptor Tyr kinase where they are phosphorylated. The target substrates phosphorylated by the receptor Tyr kinases are proteins with a common 100 amino acid domain called SH for src homology, based on structural homology to another cytoplasmic protein, Src. Src is an intracellular Tyr kinase activated when it binds through 2 SH domains to an autophosphorylated receptor Tyr kinase. Specifically, the SH2 domain has been shown to bind tyrosine-phosphorylated peptides. These domains target proteins to the autophosphorylated receptor Tyr kinase. Many proteins involved in signal transduction have SH2 domains. Some of these proteins also have catalytic domains with kinase activity. Others have phosphatase, transcription factor. or scaffolding domains.
Figure \(\PageIndex{20}\) shows the hormone-depended dimerization of RTKs, their autophosphorylation, and the recruitment of proteins with SH2 domains.

It is easier to envision how a GPCR is activated by binding its target hormone than for RTKs. GPCRs are single-chain proteins that pass through the membrane using 7-transmembrane helices. RTKs have a single transmembrane helix. The dimeric form of the RTK has some additional flexibility in the short region between the extracellular and transmembrane domains, allowing for the conformational changes necessary for the activation of the intracellular kinase domains.
The crystal structure of the full EGFR is not known given the difficulties in crystallizing membrane proteins that span the membrane with a single alpha helix. However, separate structures of the dimeric extracellular domain and the intracellular kinase domains are known.
Figure \(\PageIndex{21}\) shows an interactive iCn3D model of the dimeric extracellular and transmembrane domains of the epidermal growth factor receptor (3NJP).
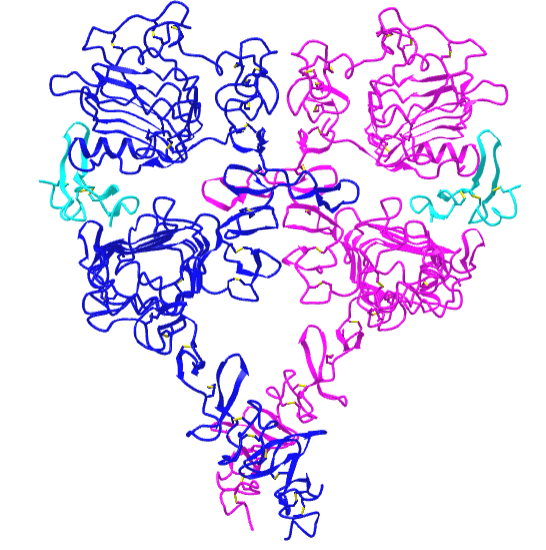
The two EGFR are shown in blue and magenta. The two bound EGFs are shown in cyan.
Figure \(\PageIndex{22}\) shows an interactive iCn3D model of a dimer of the intracellular dimeric EGFR kinase domains in complex with an ATP analog-peptide conjugate (2GS6)
.png?revision=1)
The two EGFR kinase domains are shown in cyan and magenta. The ATP analogs in each domain (spacefill) are thiophosphoric acid O-((adenosyl-phospho)phospho_-S-acetamidyldiester. The peptide substrates (green stick) are 13mers with a tyrosine (sticks, labeled Y, minus the OHs) connected to the ATP analog.
We've introduced the essential cell membrane players in signal transduction.
- GPCRs which are not enzymes but which activate the bound heterotrimer G prortein Gα subunit, which then can activate or inhibit the integral membrane protein adenylate cylase or activate the membrane-bound enyzme protein kinase C. These enzymes produce second messengers cAMP (adenylate cyclase) and diacylglycerol (DAG)and IP3 (phospholipase C)
- Receptor tyrosine kinases, ligand-activated receptor kinases, which can,on ligand-induced dimerization, autophosphorylate themselves or other target protein in the cell.
In the next chapter section we will explore the next downstream effects in signaling, mediated by the second messengers cAMP, DAG and IP3 and the substrates phosphorylated by the ligand-active receptor tyrosine kinases.