
4.11: Biomolecular Visualization - Conceptions and Misconceptions
- Page ID
- 26182


Structure determines everything in biology and chemistry. Since you learned to represent molecules with Lewis structures, it's been drilled into you that the structure of a molecule determines its physical and chemical properties. Physical properties would include melting points, boiling points, and solubility. In contrast, chemical properties include acid/base, redox, precipitation, and general chemical reactivity determined by the presence of Lewis/Brønsted acids/bases and nucleophiles/electrophiles. More modern analyzes of reactivity would include molecular orbital theory descriptions of bonding.
What makes chemistry and its fundamentally interconnected fields of biology and biochemistry so difficult to many is that we can't see molecules but make inferences from data (x-ray crystallography, NMR spectroscopy, and cryo-EM) about the structure of a molecule (atom type, atom/bond connectivity, and geometry). As the molecules get bigger (consider the muscle protein titin, also called connectin, with a molecular weight of around 3.8 million), we must use computer visualization to understand the structure and infer from it the resulting function and activity of the protein. As with small molecules, we can render the molecule in different ways to better understand various attributes of the molecule that confer function and activity. We ask students to view a biomolecule and infer its properties from the rendering without giving devoted attention and instruction as to how to do that.
Small molecules
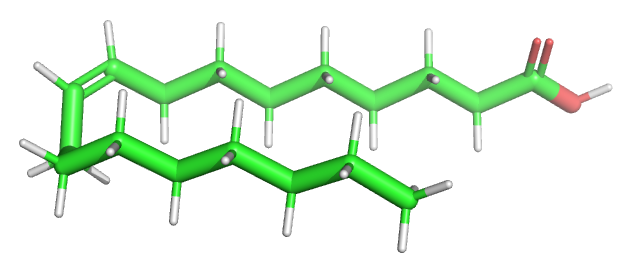
Let's start with a small molecule like oleic acid, a long chain carboxylic acid with 18 carbon atoms and one cis (Z) double bond between carbons 9 and 10 as shown in Figure \(\PageIndex{1}\).
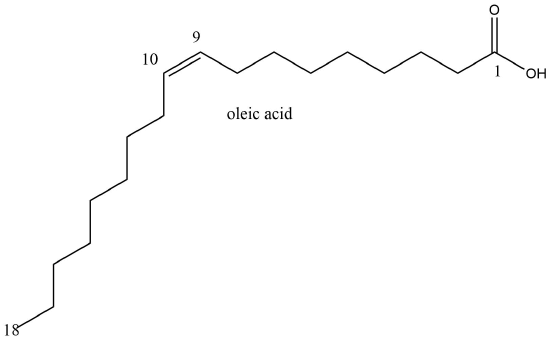
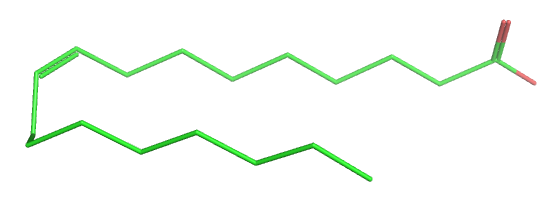
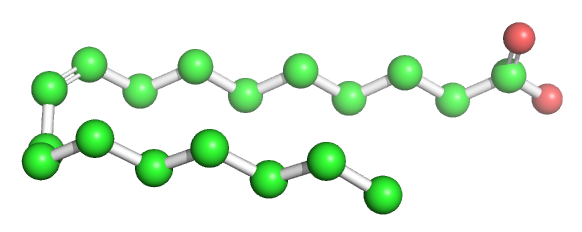
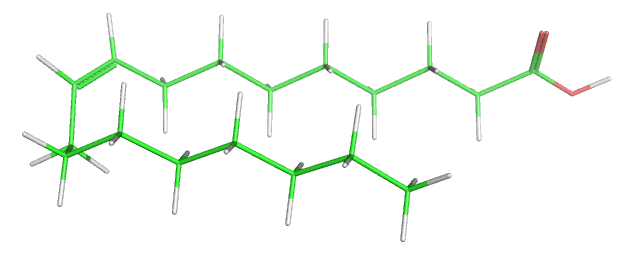
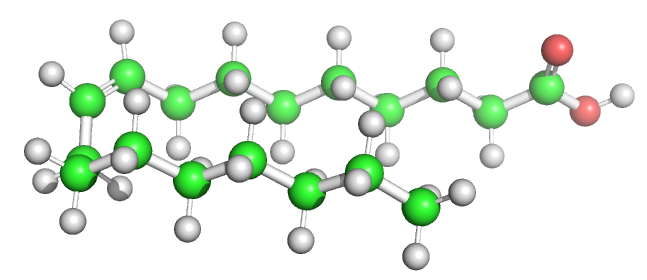
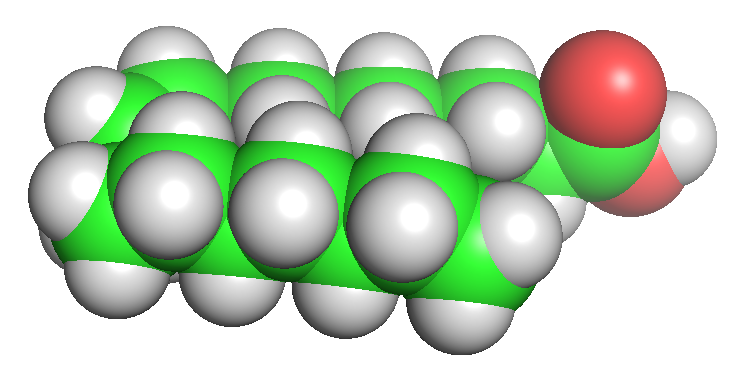
Table \(\PageIndex{1}\) below shows multiple ways to render the molecule. Each rendering offers insight in the function/activity of the molecule but might at the same time leave students with difficulties in interpreting them and also reinforce or install misconceptions. Each representation below shows the very same molecule. The top row shows representations without H atoms, which the bottom row shows them.
| line | stick | ball sphere | sphere | surface |
 |
 |
 |
 |
 |
 |
 |
 |
 |
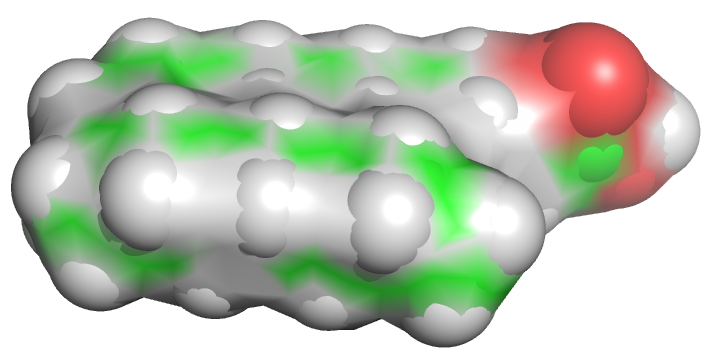 |
Table \(\PageIndex{1}\):Different renderings of oleic acid
Here are some important things to remember about biomolecular structures including both small and large molecules:
- Structures obtained using x-ray crystallography are constructed from relative electron densities calculated from diffraction patterns. Computer programs calculate the structure based on these electron density maps and known bond lengths, bond angles, atom types. Most structures used in this book are found in the Protein Data Bank. The structures seen in computer models are visualized data and can contain mistakes (missing atoms, steric conflicts, wrong atoms), although structural refinement techniques minimize such problems.
- Structures derived from X-ray and cryomicroscopy analyzes are static structures and represent only one of a large ensemble of possible conformational structures. As you learned from the study of simple molecules in organic chemistry, bond lengths and angles changes can change within molecules. Bonds connecting two atoms can stretch, angles connecting three atoms can bend, and the torsional angle around the center bond in a four-atom, three-bond system can rotate to form eclipsed and staggered (gauche and anti) conformers.
- PDB structures obtained by x-ray crystallography contain no H atoms as they are too small and contain too few electrons to diffract/scatter x-rays. So get used to adding them in your mind when you see a structure. Programs are used to calculate and show H bonds between a slightly positive H atom on an O or N atom in a protein, and another slightly negative Os or Ns on the same or different molecule. The H bonds are often shown between N and O atoms. You should look at the atoms involved and the distance between them and visualize a hydrogen atom connected to one of them. Figure \(\PageIndex{2}\) shows an example of an H bond between two base pairs in a DNA molecule.

- Double bonds or likely resonance structures are not typically shown in PDB structures.
- Line, ball and stick, and stick renderings are useful for showing connectivity between atoms and bond angles. However, they are not particularly useful in showing how atom size might affect the molecules' structure and properties. This type of information is better shown when spacefill renderings that show the sizes of the atoms (based on their Van der Waals radii) is used, or when the surface of the molecule, calculated from contact surface created between the van der Waals surface of the atoms and a rolling probe (often an O atom mimicking water) is displayed.
Large molecules
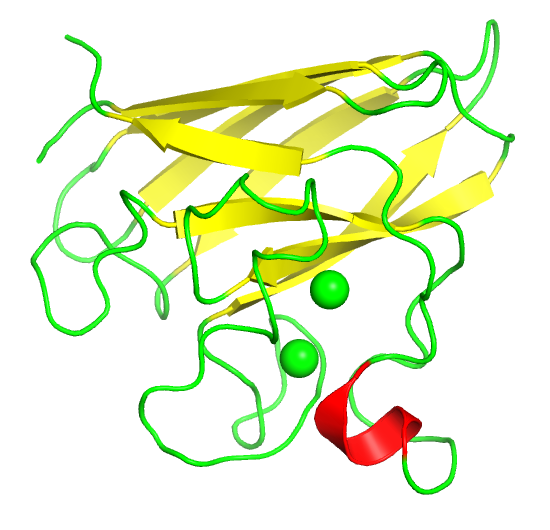
As molecules get bigger, line, stick, and ball and stick renderings are increasingly useless. New ways of visualizing the structural features of the molecules become needed. The importance of multiple renderings to clarify structure/function relationships becomes apparent when you wish to understand protein structure. Various renderings of the protein superoxide dismutase (2sod) are shown in Table \(\PageIndex{2}\) below.
| line | stick | cartoon | sphere | surface |
 |
 |
 |
 |
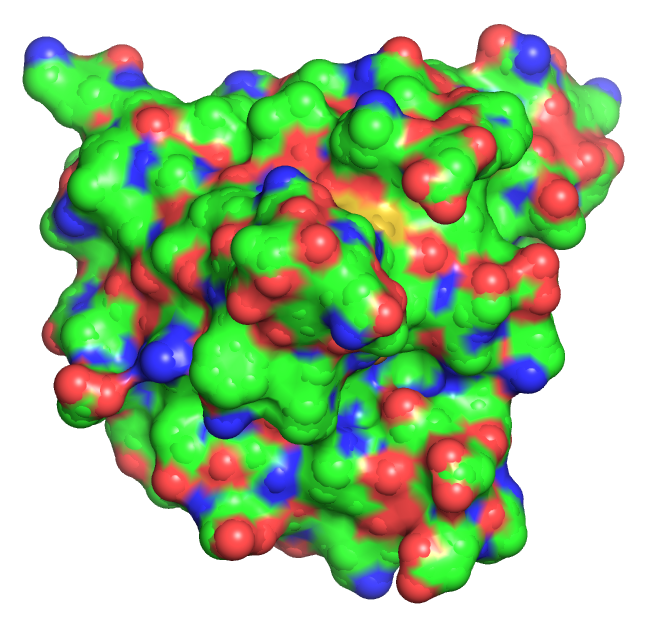 |
Table \(\PageIndex{2}\): Multiple renderings of the protein superoxide dismutase
- The same features and limitations described above for small molecules apply to large ones. It is best to leave out most of the atoms and use mixed renderings within a single display to reveal important structural feature of the biomolecule. The cartoon rendering of superoxide dismutase shows one tiny alpha helices (red) and many beta strands (yellow). All side chain and backbone atoms have been removed. The green line is the trace through the backbone of those amino acids not involved in secondary structure. The Cu and Zn ions are shown as spheres.
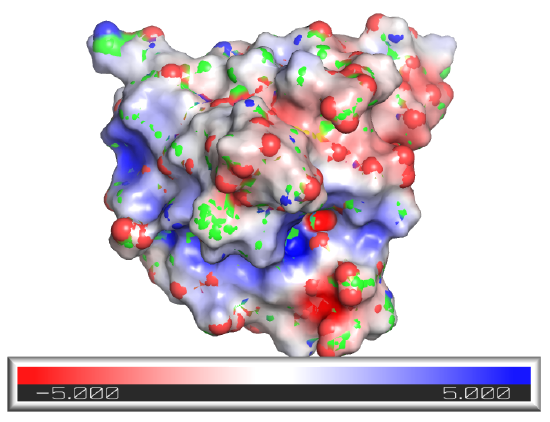
Another type of surface rending, the electrostatic potential surface, is beneficial. Figure \(\PageIndex{3}\) shows the electrostatic potential surface of superoxide dismutase taken from two different angles after simply rotating the protein.
 |
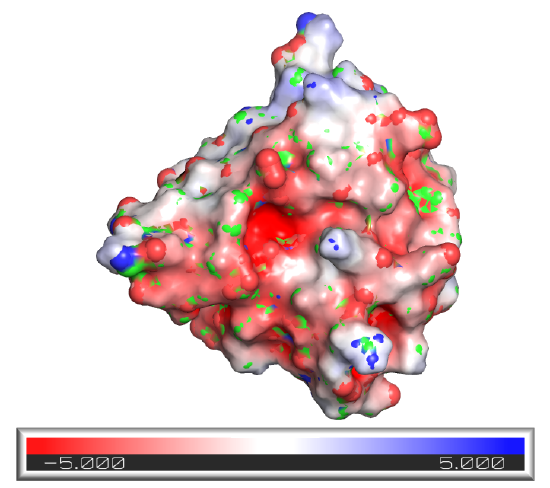 |
Figure \(\PageIndex{3}\): Electrostatic potential surface of superoxidase dismutase from two perspectives
The red represents minimal (most negative) potential. This part of the structure would be enriched in slightly negative Os and Ns or fully negative Os (i.e. have the highest electron density). The representesents the positive potential, centered in areas of the surface containing slight or full positive charge (I,.e.the lowest electron density). This enzyme binds superoxide, O2-, a toxic free radical reduction product of dioxygen, \(\ce{O2}\). It catalyzes this reaction:
\[\ce{2 O2^{-} + 2H^{+} → O2 + H2O}. \nonumber \]
The enzyme can effectively scavenge superoxide in its vicinity as the negative superoxide is drawn into the active site with the \(\ce{Cu}\) and \(\ce{Zn}\) atoms by the positive potential surrounding the active site, enhancing the normal diffusion encounter rate of the reactant with the enzyme pocket.
Figure \(\PageIndex{4}\) shows an interactive iCn3D model of the electrostatic potential of superoxidase dismutase (2sod).
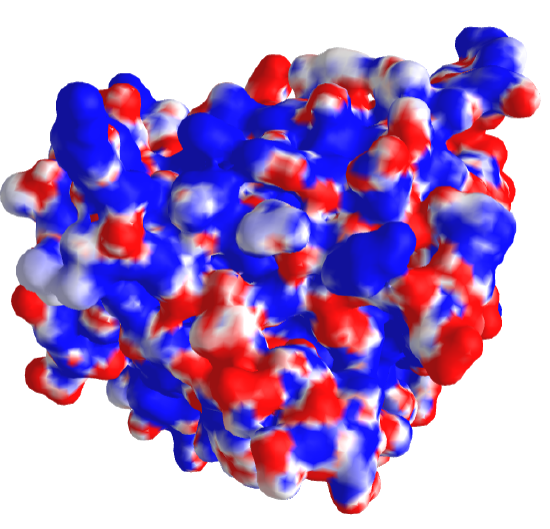
 Figure \(\PageIndex{4}\): Electrostatic potential of superoxidase dismutase (2sod) (Copyright; author via source).
Figure \(\PageIndex{4}\): Electrostatic potential of superoxidase dismutase (2sod) (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...QQy2hAXehWLBW8