
List of Figures
- Page ID
- 18932

List of Figures
Figure 1.1: Tetracycline is a natural antibiotic produced by soil bacteria. It is a polycyclic aromatic compound.
Figure 1.2: Bacteria render antibiotics ineffective using one of four strategies: they degrade the antibiotic (A); they alter the target through mutation (B); they block the entry of antibiotics (C) or pump them out of the cell with the help of efflux pumps (D).
Figure 1.3: A model depicting the conformational change of the ykkCD toxin sensor upon tetracycline binding. When tetracycline levels rise to a critical threshold the ykkCD sensor binds to tetracycline and undergoes a structural change that permits production of the MDR pump.
Figure 1.4: Schematics depicting specific target recognition by a macromolecule. The specific target is able to form all noncovalent interaction with the sensor leading to a low KD value for the sensor-target complex (top). Any other molecule is rejected, because not all noncovalent interactions can be formed (bottom).
Figure 1.5: Schematics depicting how to evaluate the results of the ykkCD RNA mutagenesis study. If the nucleotides mutated were not important for tetracycline recognition then all important noncovalent interactions still form between tetracycline and the sensor and thus a low KD value is measured (top). If the nucleotides mutated were important for recognition then not all noncovalent interactions are able to form between the mutant sensor and tetracycline. As a result a high KD value is measured (bottom).
Figure 2.1: Evaluation of ykkCD sensor RNA mutants using binding affinity assays: If 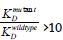 (higher KD value) the nucleotide(s) mutated were important for tetracycline recognition. If
(higher KD value) the nucleotide(s) mutated were important for tetracycline recognition. If 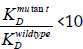 (10 (low KD value) the nucleotides mutated were not important for tetracycline recognition.
(10 (low KD value) the nucleotides mutated were not important for tetracycline recognition.
Figure 2.2: Global alignment versus local alignment: Global sequence alignment aims to find a GenBank sequence that shows significant overall similarity to the query sequence. Local sequence alignment attempts to find a GenBank sequence that shows discrete regions of significant similarity.
Figure 3.1: Schematics of the first PCR amplification cycle. Note that the desired DNA sequence is not generated yet.
Figure 3.2: Schematics of the second cycle of PCR amplification. Note that the desired product is generated.
Figure 3.3: Schematics of cutting and pasting: During conventional cloning (cutting and pasting) both the plasmid vector and the amplified DNA has to be cleaved with the same pair of restriction enzymes and ligated together.
Figure 3.4: Schematics of Quickchange mutagenesis.
Figure 4.2: Polynucleotide structure: Nuclotides in a nucleic acid are held together by the phosphodiester bond; the chain goes in the 5’ to 3’ direction.
Figure 4.3: Mechanism of polymerization: The 3'-OH group of the growing nucleic acid chain acts as a nucleophile and attacks the alpha phosphorous of the incoming nucleotide triphosphate to catalyze formation of the phosphodiester bond.
Figure 4.4: The newly synthesized nucleic acid has complementary sequence and opposite chain direction than the template
Figure 4.5: Schematic of a PCR cycle. Note that each step takes place at a different temperature so PCR machines have to be able to change temperature quickly and precisely.
Figure 5.1: Supercoiled DNA (left) is more compact than circular DNA (right) of the same molecular weight (Courtesy of Dr. Jack Griffith).
Figure 5.2: Migration of DNA on an agarose gel depends on shape and size. Shorter DNAs migrate faster than longer DNAs. Note the separation of DNAs in the MW marker.
Figure 5.3: Nucleic acid visualization using EtBr: EtBr is aromatic. As a result, it chelates with nucleobases (left). EtBr treated nucleic acids light up when shined with UV light, because EtBr is fluorescent in the ultraviolet range (right).
Figure 6.1: Mechanism of action of restriction endonucleases: Restriction endonucleases cleave dsDNA with high specificity. They use a metal-ion bound water molecule as nucleophile to catalyze cleavage of the phosphodiester bond.
Figure 6.2: Restriction endonucleases and their target sites have complementary twofold symmetry. The monomers of the restriction endonuclease homodimer are shown in green and red; the dsDNA template is purple (PDB ID 1ERI).
Figure 6.3: The cognate DNA is bent by the restriction endonuclease to position the scissile bond into the active site for cleavage.
Figure 6.4: The extensive H-bonding between the restriction endonuclease and its cognate DNA substrate provides the energy for DNA bending. DNA bending is essential for catalysis (see text).
Figure 6.5: The success of restriction endonuclease cleavage is judged by resolving the reaction mix on an agarose gel.
Figure 7.1: Chemistry of polymerization. The 3'-OH group of the growing nucleotide chain acts as nucleophile to attack the α-phosphorous of the incoming NTP to catalyze formation of the phosphodiester bond.
Figure 7.2: The transcription bubble: After separating the dsDNA template the RNA polymerase moves from 3' to 5' direction on the DNA template to synthesize RNA. The RNA sequence is the same as the top DNA strand, but each T is replaced with U.
Figure 7.3: Schematics of a bacterial promoter. Note that the two specific sequences are required to be positioned at a given distance from each other for optimal polymerase binding.
Figure 7.4: T7 RNA polymerase transcription initiation complex (PDB ID 1QLN): The T7 RNA polymerase recognizes a hairpin-shaped promoter. The RNA chain grows in the 5’ to 3’ direction. The RNA sequence is the same as the top DNA strand except each T is replaced with U.
Figure 7.5: Schematics of a terminator stem: The G/C rich stem is stable and stalls the polymerase; the A/U rich tail releases the nascent RNA.
Figure 8.1: Denaturing polyacrylamide electrophoresis of RNAs. (A) RNA molecules are separated by molecular weight. The DNA template in the unpurified sample migrates on top, because of its larger molecular weight. (B) Urea is a chaotrop that unfolds RNAs by H-bonding with nucleobases.
Figure 8.2: Polyacrylamide gel electrophoresis. The polyacrylamide medium is generated by free-radical polymerization. The ratio of acrylamide: methylenebisacrylamide determines the pore size of the gel.
Figure 9.1: The KD value is the sensor RNA concentration that forces 50% of the tetracycline to form the tetracycline-ykkCD RNA complex.
Figure 9.2: Fluorescence is a natural phenomenon (top). Fluorescent compounds emit light that is lower in energy, and of higher wavelength, than the light absorbed (bottom).
Figure 9.3: Jablonski diagram illustrates the electronic states of a molecule undergoing fluorescence. The electron is excited from the ground state to a higher energy excitation state. From there it releases energy through nonradiative decay then emits light (fluorescent light) to return to the ground state.
Figure 9.4: Diagram of fluorescent binding assay. Once tetracycline is bound to the sensor its natural fluorescence decreases leading to fluorescent quenching.
Figure 9.5: Determination of KD value. The amount of quenching is plotted against the sensor RNA concentration. The KD value is the sensor RNA concentration where 50% of quenching is achieved.
Figure 9.6: Schematics illustrating how to interpret the results of the binding affinity assays: Mutant 1 binds strongly to tetracycline, thus we conclude that the nucleotides changed were not essential for tetracycline recognition. In contrast, mutant 2 did not bind strongly to tetracycline. This means the nucleotides changed were probably important for tetracycline recognition.
Figure 9.7: Example of data analysis. The binding affinity of the mutant sensor RNA-tetracycline complex is elevated (upper panel) compared to that of the wild-type sensor RNA tetracycline complex (lower panel). Elevated KD value corresponds to weakened binding to tetracycline. Thus we conclude that the mutated nucleotides were important for tetracycline recognition.
Figure 9.8: Saturation curve depicting binding of a ligand to a receptor. KD value indicates the ligand concentration at which the binding site on a particular protein is half occupied.
Figure 9.9: Saturation curve depicting binding of tetracycline to the sensor RNA. KD value indicates the ligand concentration where 50% of the sensor RNA is bound with tetracycline.
Figure 9.10: Linear representation of a saturation curve. The KD value may be determined in two different ways: (1) the slope of the graph equals the KD value or (b) the x-intercept is -1/KD.
Figure 9.11: B. subtilis ykkCD sensor RNA secondary structure: Color coding is as follows: red, 100% sequence conservation; blue, >80% sequence conservation and black, no significant sequence conservation.
Figure 10.1: An energy diagram showing the energy transfer during a fluorescent process. Absorbed light excites an electron to a higher energy orbital. Some of the absorbed energy is lost to molecular vibrations. The remainder of the absorbed energy is released as light when the excited electron returns to the ground state.
Figure 10.2: Block diagram showing the major spectrofluorometer components used to measure an emission spectrum. Fluorescent light is emitted from all sides of the solution containing fluorescent molecules and is measured at right angles to the excitation light to minimize background interference.
Figure 10.3: A block diagram showing the major spectrofluorometer components used to measure an excitation spectrum. Note that a typical research-grade spectrofluorometer has two monochromators, one for excitation light and one for emission light.
Figure 10.4: Energy diagrams that qualitatively compare the tyrosine fluorescence with the tryptophan fluorescence. The tyrosine fluorescence (shown on the left) is higher in energy (shorter wavelength) than the tryptophan fluorescence (shown on the right). Once excited, tryptophan loses more energy to vibrations than does tyrosine.
Figure 10.5: Common graphical representations of ligand binding to a protein. In both graphs the dependent variable is a function of [PL] while the independent variable is [L]. The graph on the right plots the ratio 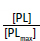 . When plotted this way, the Y-axis varies from 0 to 1.
. When plotted this way, the Y-axis varies from 0 to 1.
Figure 10.6: Graphical representation of levofloxacin binding to human serum albumin: The independent variable is the levofloxacin concentration. The dependent variable is fluorescence quenching which is equal to .
Figure 10.7: Three overlaid fluorescence spectra illustrating a typical spectrum and the impact of a blue shift or a red shift.
Figure 12.1: A schematic of the acetylcholinesterase active site showing the electron movement during the initial nucleophilic attack of acetylcholine: The alcohol acts as a nucleophile attacking the ester carbonyl carbon. This alcohol is a strong nucleophile because the adjacent imidazole ring changes the alcoholic hydroxyl group to an alkoxide. In other words, the imidazole acts as an especially strong base. It can act in this way, because of the adjacent carboxylate negative charge.
Figure 12.2: A schematic of the acetylcholinesterase active site showing the covalent intermediate formed following the initial nucleophilic attack: Note that the negative charge that forms on the carbonyl oxygen is stabilized by hydrogen bonding with “-NH” groups. Once this covalent intermediate forms, choline rapidly leaves the active site and water enters to complete the reaction.
Figure 13.1 : A Michaelis-Menten plot graphs steady-state reaction velocity as a function of substrate concentration: Each data point on the plot represents one assay. This plot reports data from seven assays.
Figure 14.1: Schematic showing of the connections needed to build a phosphatidylcholine.
Figure 14.2: A chromatogram showing the separation of molecule A from molecule B: The x-axis is measured as retention time, the time a solute spends on the chromatography column after the sample is injected. The void time is the fastest time a solute pass through the column, i.e., the solute does not stick to the stationary phase at all. “(Reproduced with permission, from M. Ferrer, O.V. Golyshina, F.J. Plou, K.N. Times and P.N. Golyshin, (2005), Biochemical Journal, 391(2) 269-276. © the Biochemical Society)
Figure 14.3: Structures of the phosphatidylcholines to be separated in the experiment.