
Homework Problems - Literature Learning Module: Steroidogenic Acute Regulatory Protein - KEY
- Page ID
- 9977

Research Paper: A pH-dependent Molten Globule Transition Is Required for Activity of the Steroidogenic Acute Regulatory Protein, StAR. Bo Y. Baker, Dustin C. Yaworsky, and Walter L. Miller J. Biol. Chem., Vol. 280, Issue 50, 41753-41760, December 16, 2005
Steroids synthesis requires cholesterol, which get converted to pregnenolone by an enzyme in the inner mitochondrial membrane (IM). A regulatory protein, which we will call SR, is found in the outer mitochondrial membrane (OM) and is involved in cholesterol movement from the OM to the IM. Mutations in SR can cause congenital lipoid adrenal hyperplasia, a potentially lethal disease. The protein is synthesized in the cytoplasm as a 37,000 MW pre-protein, whose N terminal "leader sequence" targets SR to the mitochondria. Upon translocation of the protein to the IM, the leader is cleaved by proteolysis and is removed from the final active protein. The C-terminus must be critical for it's function since mutations in this region causes the hyperplasia discussed above.
Models of SR suggest that it is similar to related protein that have a β-barrel structure with a hydrophobic binding pocket which can bind cholesterol. If the first 62 N-terminal amino acids are removed from the precursor ("preprotein"), the protein (N-62 SR) retains activity. When added to LUVs with a lipid composition similar to the OM, only the C-terminal alpha helical end of the protein is protected from added proteases, suggesting that that end inserts into the OM, effectively tethering the protein to the OM. Using spectral analysis and proteolysis of the protein, it appears that the protein may adopt a molten globule state at both low pH or when bound to a LUV. A conformation change appears to be induced when the protein interacts with protonated head groups of the LUV. The C-terminal helix is more protected from proteolysis at pH 4.0 compared to 6.5. Modeling studies based on x-ray structures of homologous proteins show that the access channel to the hydrophobic binding site in the protein is not large enough to pass cholesterol. These results show that the protein must change conformations to elicit its biological activity. The experiments described below were done to determine if a conformational change occurs in the protein on interaction with lipids, and whether a molten globule state of the protein is formed.
A model structure of N-62 SR is shown below in Figure 1A. It has a tightly folded N-terminal domain and a more loosely folded C-terminal domain. A molecular dynamics simulation of the protein under 3 different protonation states was done in a bath of 9631 water molecules, in order to simulate possible conformational changes in the protein. The first charge state was defined to be neutral, simulating pH 7.0. Next they changed the protonation state of amino acids to reflect expected charges under acidic (pH 4) condition, which might promote a change to the molten globule state. In a last trial, they left the majority of the protein under neutral pH condition, but changed just the charge state of the C-terminal helix to acidic (pH 4.0) conditions.
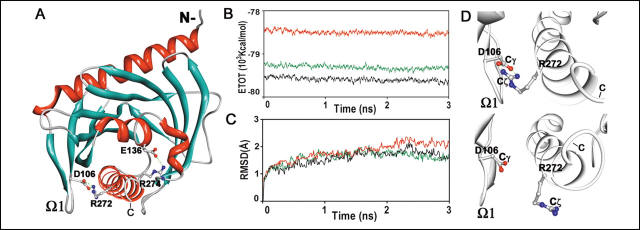
FIGURE 1.
Modeling and molecular dynamics of N-62 SR. A, ribbon diagram of the model of N-62 SR; the salt bridges between Asp (D)106 and Arg (R) 272 and between Glu (E)136 and Arg (R) 274 are shown as ball-and-stick representations (white, carbon; blue, nitrogen; red, oxygen). B, total energy levels of MD simulations as a function of time. Black, neutral; red, acidic; green, acidified C-helix. C, root mean square deviation (RMSD) of ![]() -carbons during MD of the initial N-62 SR structure. D, images of Asp-106-Arg-272 after 2800 ps of MD under neutral (top) and acidified C-helix conditions (bottom). The carboxyl carbon (Cγ) of Arg-106 in the ω1 loop (left) is bonded to two oxygen atoms, (Oδ1 and Oδ2), and the guanidino carbon (C
-carbons during MD of the initial N-62 SR structure. D, images of Asp-106-Arg-272 after 2800 ps of MD under neutral (top) and acidified C-helix conditions (bottom). The carboxyl carbon (Cγ) of Arg-106 in the ω1 loop (left) is bonded to two oxygen atoms, (Oδ1 and Oδ2), and the guanidino carbon (C![]() ) of Arg-272 in the C-helix (right) is covalently bonded to two nitrogen atoms (N
) of Arg-272 in the C-helix (right) is covalently bonded to two nitrogen atoms (N![]() 2 and N
2 and N![]() 1). Under neutral conditions, Arg-106 forms two hydrogen bonds to Arg-272 (Oδ1-Nω2 and Oδ2-Nω1), but under acidified C-helix conditions, the ω1 loop and C-helix movement and bonding are disrupted.
1). Under neutral conditions, Arg-106 forms two hydrogen bonds to Arg-272 (Oδ1-Nω2 and Oδ2-Nω1), but under acidified C-helix conditions, the ω1 loop and C-helix movement and bonding are disrupted.
1. Which amino acids would be protonated on the shift from neutral to acidic (pH 4.0) conditions. Explain why the authors ran a simulation with just the C-terminal helix in the acidic protonation state.
Ans: The amino acids that are deprotonated by neutral pH would most likely be His (with a pKa of 6 hence partially deprotonated) and Asp and Glu, with pKas around 4, whose side chains carboxylic acids would be negatively charged. at neutral pH. It is the C-terminus that is protected from proteolysis when the protein is bound to the membrane, suggesting that is might be buried in the bilayer. That is unlikely to happed if the C terminus contains a negative charge, which would be minimized at pH 4.0
2. Compare the energies of the neutral and acidified strucutures (1B) and the rigidity of the two structures in 1D. Which one is more compact? Flexible? Higher in energy? Which would be more consistent with a molten globule conformation? Do these structures support the notion that the protein might interact with the membrane through its C-terminal helix?
Ans: It appears that the acid structure is less rigid, more open, consistent with the notion of greater conformational flexibility necessary for insertion of this end into the membrane. The acidified versions are also higher in energy, suggesting again a state not in the global minimum and which could be further stabilized by interactions with membrane.
3. Snapshots of molecular dynamics simulations were taken every 100 picoseconds, and the distance between the Cγ of Asp (D)106 in the ω loop and the guanidino group of Arg (R) 272 on the C-terminal alpha helix was determined and represented graphically below in Figure 2C (top panel) below (same color coding as before). The bottom panel in 2C shows the distance between Asp (D)136 and Arg (R) 274.
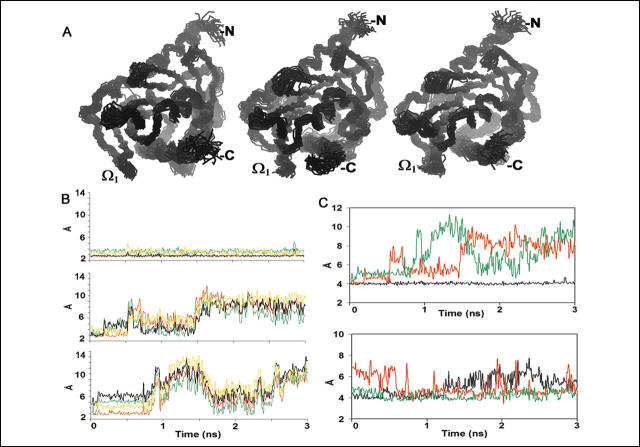
FIGURE 2.
Movement of the StAR molecule during molecular dynamics. A, C![]() superposition of conformations from MD snapshots every 100 ps over the 3-ns simulations at neutral (left panel), acidic (middle panel), and acidified C-helix (right panel) conditions. The protein is dynamic, particularly in the distal ends and the loop regions, but the overall structure is well maintained at neutral and acidic pHs. At low pH, conformation transitions are particularly prominent at the Ω1 region. B, interatomic distances (in �) between Arg-272 and Asp-106 as a function of time during molecular dynamics simulations. The carboxyl carbon (Cγ) of Arg-106 in the Ω1 loop is bonded to two oxygen atoms, termed Oδ1 and Oδ2, and the guanidino carbon (Cζ ) of Arg-272 in the C-helix is covalently bonded to two nitrogen atoms, termed Nω1 and Nω2. The distances are plotted between these atoms and shown as follows: in red,Oδ1/Nω2; green,Oδ1/Nω1; black,Oδ2/Nω1; and yellow,Oδ2/Nω2. Under neutral conditions, the Oδ1/Nω2 and Oδ2/Nω1 distances are 2.8 � 0.1 �, favoring hydrogen bonding; pairings of Oδ1/Nω1 and Oδ2/Nω2 are less likely. Upper panel, neutral; middle panel, acidic; lower panel, acidified C-helix. C, interatomic distances during MD. Top, distance (in �) between Cζ in Arg-272 and Cγ in Asp-106. Bottom, distance (in �) between Cζ in Arg-274 and Cδ in Glu-136. Black, neutral; red, acidic; green, acidified C-helix.
superposition of conformations from MD snapshots every 100 ps over the 3-ns simulations at neutral (left panel), acidic (middle panel), and acidified C-helix (right panel) conditions. The protein is dynamic, particularly in the distal ends and the loop regions, but the overall structure is well maintained at neutral and acidic pHs. At low pH, conformation transitions are particularly prominent at the Ω1 region. B, interatomic distances (in �) between Arg-272 and Asp-106 as a function of time during molecular dynamics simulations. The carboxyl carbon (Cγ) of Arg-106 in the Ω1 loop is bonded to two oxygen atoms, termed Oδ1 and Oδ2, and the guanidino carbon (Cζ ) of Arg-272 in the C-helix is covalently bonded to two nitrogen atoms, termed Nω1 and Nω2. The distances are plotted between these atoms and shown as follows: in red,Oδ1/Nω2; green,Oδ1/Nω1; black,Oδ2/Nω1; and yellow,Oδ2/Nω2. Under neutral conditions, the Oδ1/Nω2 and Oδ2/Nω1 distances are 2.8 � 0.1 �, favoring hydrogen bonding; pairings of Oδ1/Nω1 and Oδ2/Nω2 are less likely. Upper panel, neutral; middle panel, acidic; lower panel, acidified C-helix. C, interatomic distances during MD. Top, distance (in �) between Cζ in Arg-272 and Cγ in Asp-106. Bottom, distance (in �) between Cζ in Arg-274 and Cδ in Glu-136. Black, neutral; red, acidic; green, acidified C-helix.
Referring to the location of the amino acids structure shown in Fig 1A, and the data in Fig 2C, which part of the protein is likely to change to promote access of cholesterol to sterol binding pocket in the protein?
Ans: To visualize the movement of the C-helix, we superimposed images of the molecule obtained every 100 ps, and we plotted the distances between key pairs of atoms during MD. The distance between the carboxyl carbon (Cγ) of Asp-106 in the Ω1 loop to the guanidino carbon (Cζ) of Arg-272 remained stable at 4 � under neutral conditions, whereas under either acidic or acidified C-helix conditions, this distance fluctuated dramatically, reaching >10 � (Figure 2C, top). This indicates that the C-helix and Ω1 loop move dramatically with respect to one another, opening and closing the SBP. Similar hydrogen bonding was seen between Glu-136 and Arg-274 in the static model at neutral pH. This distance varied from 4 to 6 � during MD at neutral pH but was relatively more stable under acidic conditions, particularly when only the C-helix was acidified (Figure 2C, bottom). Thus, acidic pH had little effect (or even a stabilizing effect) on the distance between Arg-274 in the C-helix and Glu-136 in the short α2 helix that contributes to the roof of the SBP. Therefore, cholesterol is more likely to reach the SBP via a space between the Ω1 loop and the C-helix than between the Ω3 loop and the C-helix.
In an attempt to run an additional control for their "in silico" molecular dynamics experiment, the authors found two pairs of amino acids with the amino acids in each pair being approximately 4 angstroms from each other in the C helix and adjacent loop. The two pairs were Ser 100/Ser 261 and Asp 106/Ala 268. They changed them to Cys and created "in silico" the following mutation pairs (in single letter code): S100C/S261C and D106C/A268C. Note: DA mutant places the C268 closer to the end of the C helix than in the SS mutant, where the C is position 261
4. Why did they believe that changes these amino acids to Cys would not dramatically change the folding of the protein?
ANS: Cys are similar in size to Serine, Asp, and Ala and did not add a lot of "hydrophobicity" to the protein which may have contributed to misfolding.
5. Energy minimization results showed that the Cα backbone of the C-terminal helix or Ω loop did not change. What would the likely effect be on the local structure in the vicinity of the pair? How would the molecular dynamics simulation in the top panel of 2C change?
Ans: Since the overall energy was similar, the mutants and WT were probably similar in structure. However, the mutants would have a much greater rigidity in the C terminus region, which should be evident in reduced motion in MD simulations. Why? They engineered disulfides into the protein in that region.
The mutants described above where made using site-specific mutagenesis, and SDS-polyacrylamide gels run.
6. Why did the mutants (DA) and SS migrated further on the gel than the wild type (WT) unmutated proteins?
Ans: The mutants were more conformational restricted (due to the engineeredd disulfide bonds) and upon denaturation with SDS would not denature as completely compared to WT (unless the disulfides were first reduced). Hence they would be more compact and migrate faster in the PAGE gel.
7. When the mutant proteins were treated first with diothiothreitol (DTT) before electrophoreis, they comigrated with the WT proteins. When the mutant proteins were treated with diamide (oxidizes sulfhydryls to disulfides), their migration was not affected. The structures of DTT and diamide are shown below. Explain the gel results.
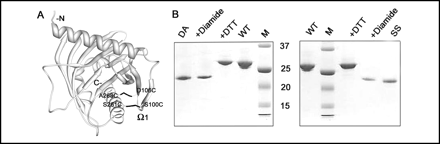
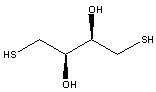
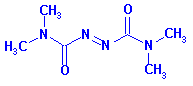
FIGURE 3.
Characterization of the disulfide mutants. A, model of N-62 SR showing the locations of the SS mutant replacing Ser-100 and Ser-261 and the DA mutant replacing Asp-106 and Ala-268 with Cys. Both mutants are shown in a single molecule, but each was prepared separately. Note that the perspective is from "behind" the molecule, as compared with Figure 1A. B, SDS-PAGE gel of the SS (right) and DA (left) mutants. Lane designations in each gel are as follows: M, molecular weight standards; WT, wild type N-62 SR; SS and DA, untreated mutant proteins; diamide, mutant plus diamide; DTT, mutant plus DTT.
Ans: DTT would reduce the disulfides in the mutant proteins back t o free Cys, which would cause the mutant proteins to comigrate with the WT protein. The diamide would not affect the protein since no free Cys were present in the WT protein since they were present already as disulfides.
8. The amino acid sequence, as derived from the DNA sequence, shows that the WT N-60 protein has 3 Cys and the two mutants (DA and SS mutated as above to contain two Cys) each have 5. The amount of free Cys in each protein was determined using Ellman's reagent. Draw a reaction to show how a free Cys (represented as RSH below) reacts with Ellmans' reagent. What instrument would you use to measure the reaction and quantitate the levels of Cys?
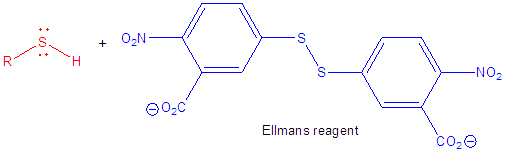
Ans: Use a spectrophotometer to measure the increase in A405 nm due to the leaving group formed when Ellman's reagent interacts with free Cys residues.
-
Reaction mechanism:
9. Computer modeling of the WT N60 SR shows no disulfides and two accessible Cys. Compare the number of Cys residues determine by Ellman's for each protein in the presence and absence of sodium dodecyl sulfate.
| Protein | # Cys - SDS | # Cys + SDS |
| WT | 2 | 3 |
| DA | 2 | 3 |
| SS | 2 | 3 |
Why? The mutants have the extra 2 Cys tied up in a disulfide, which don't react with Ellmans reagent. SDS denatures the protein exposing the buried Cys to Ellmans reagent, but does not cleave disulfides.
10. A comparison of the CD spectra and the fluorescence emission spectra of the WT protein and two mutants, SS and DA were made. Why was this done?
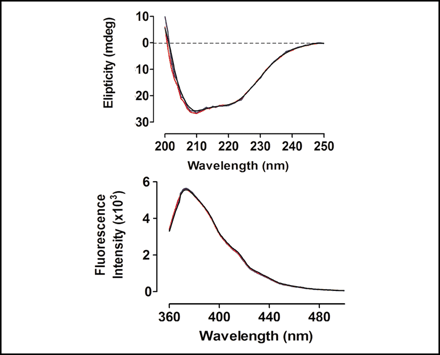
FIGURE 6.
Spectroscopic analysis. Top, far UV-CD spectra; bottom, fluorescence spectra of wild type (black), SS mutant (blue), and DA mutant (red) of N-62 SR.
Ans: To provide experimental evidence (outside of in silico experiments) that the structure of the mutants and WT were generally similar.
Next CD measurements were made on the WT, SS and DA mutants at various pH to s
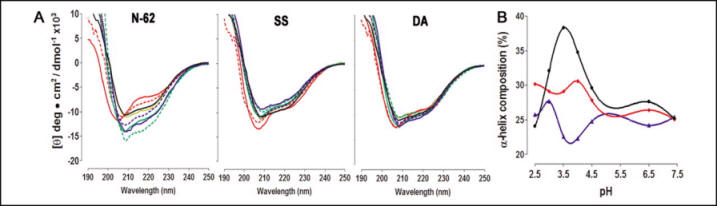
FIGURE 7.
A, far-UV CD spectra of the wild type (left), SS mutant (middle), and DA mutant (right) of N-62 StAR. Each protein was assessed at pH 2 (solid red line), pH 2.5 (dotted red line), pH 3.0 (solid green line), pH 3.5 (dotted green line), pH 4.0 (solid blue line), pH 4.5 (dotted blue line), pH 6.0 (solid tan line), and pH 7.4 (black line). B, predicted ![]() -helical content of wild-type (black), SS (blue), and DA N-62 SR (red).
-helical content of wild-type (black), SS (blue), and DA N-62 SR (red).
11. Compare the spectra in Fig 7A of the mutants, especially the DA mutant) to the WT. Also note the data in 7B. What is the overall effect on the change in the structure of the protein within in the mutants? Is all the data consistent with the idea that formation of the molten globule might play a role in the activity of the protein?
Ans: To test the effects of immobilizing the C-helix on the pH-dependent molten globule properties of StAR, we measured the CD spectrum of the wild type, SS mutant, and DA mutant at pH 2-7.5 (Fig. 7A). Analysis of these data by several algorithms showed that acidification induced a substantial increase in the α-helical content of wild-type StAR, with maximal helicity at pH 3.5, as reported previously (Figure 7B). Consistent with the overlapping spectra in Figure 6, the calculated α-helical content of the wild type and mutants was the same at pH 7.5, but the mutants showed substantially less change in α-helical content at acidic pH, with no peak at pH 3.5 (Figure 7B). Thus, the disulfide mutants prevented the generation of the principal spectroscopic hallmark of the pH-induced molten globule transition described previously.
12. Based on your conclusions and the data, rank the following proteins as to their cholesterol binding activity: WT, SS mutant, DA mutant. Explain.
Ans: StAR can bind cholesterol with 1:1 stoichiometry. Wild-type N-62 StAR bound 14C cholesterol and fluorescent NBD-cholesterol. Cholesterol binding by the SS mutant was reduced by about 50%. The engineered disulfide bond in the SS mutant is in the middle of the C-helix (S261C), which only partially restricted movement of this helix. Binding by DA was similar to the buffer background or binding by the inactive StAR mutant M144R . The DA mutant contains a disulfide closer to the carboxyl terminus (A268C), which more completely immobilized the C-helix and eliminated all measurable cholesterol binding. The cholesterol binding activity of both mutants was fully restored by treating the disulfide mutants with DTT . Similarly, wild-type N-62 StAR elicited five times more steroidogenesis from mitochondria isolated from mouse Leydig MA-10 cells than did buffer. However, the activity of the SS mutant was reduced by about half and the activity of the DA mutant was similar to the buffer background, but these two mutants recovered full activity when the disulfides were disrupted with DTT . Thus, the SS and DA mutants were fully capable of normal function when their capacity to form a molten globule was restored by cleaving their disulfide bonds.