
F9. Protein Stability and Molecular Orbitals
- Page ID
- 4772

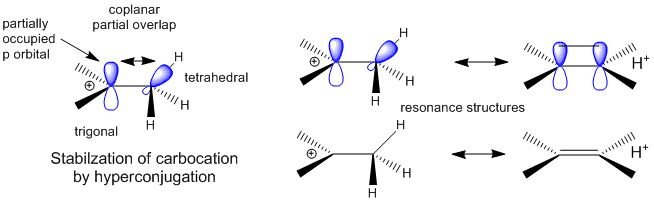
A more complete description of the energetics underlying protein structure and stability would involve quantum mechanical effects. These have not been studied in detail since quantum mechnanical calculations are difficult to perform on large molecules. Recent work has led an understanding of a new source of stability of proteins based on overlap of proximal molecular orbitals that stabilizes a protein through electron delocalization. Two common examples of electron delocalization that increase stability are resonance and hyperconjugation. Resonance involves delocalization of electrons through overlap of adjacent atomic p orbitals to form low energy molecular bonding orbitals. You may remember hyperconjugation being used to explain the relative stability of tertiary carbocations compared to secondary or primary ones. The typical explanation involves electron donation and pushing from the adjacent methyl groups which effectively decreases the charge on the carbocation. A more correct explanation involves overlap of molecular orbitals, specifically of a C-H s orbital from one of the C-H bonds of the methyl group with the partially filled p orbital on the carbon atom containing the charge as seen in the figure below.
Figure: Stabilization of Carbocations by Hyperconjugation
It should also not be surprising that orbital overlap accounts for hydrogen bonds interactions (in contrast to the more simplistic explanation of the attractions between the partial charges on hydrogen bond donors and acceptors). Bartlett et al (2010) have recently described two types of orbital overlaps involving electron delocalization that stabilize protein structures. Molecular orbital description show that two lone pairs on the oxygen (hydrogen bond donor) in amide bonds are different. One pair, ns (Figure a below) is in a nonbonding orbital with 60% s character, while the other, np (figure b below), has approximately 100% p character. Figure c below shows that a hydrogen bond between the carbonyl oxygen of the ith amino acid and the amide H of the ith+4 amino acid can be described in quantum mechanical terms as the overlap (i.e. delocalization) of the ns nonbonding orbital on oxygen and the s* antibonding N-H orbital. What about the np electrons on the carbonyl O? They found evidence for a new kind of stabilizing influence in proteins as the np orbital overlaps with the an antibonding p* orbital on the ith+1 carbonyl group (figure d below) when the two amino acids are proximal as parts of secondary structures, including alpha and 310 helices as well as in twisted b sheets.
Figure: Orbital involvement in H-bonds and n to pi* interactions in alpha helices
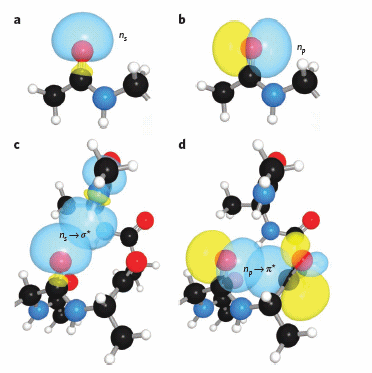
(a) s-rich lone pair of an amide oxygen. (b) p-rich lone pair of an amide oxygen. (c) ns to σ*: hydrogen bond in an a-helix. (d) np to p*: n to p* interaction in an a-helix. Reprinted by permission from Macmillan Publishers Ltd: Nature Chemical Biology. Bartlett, G. et al. n to pi* interactions in protein. 6, pg 615 (2010)
To test which phi-psi angles would allow close enough approach for np to p* interactions, the investigators performed quantum mechanical computations on possible conformations of AcAla4NHMe for which np to p* were possible (see figure A below). These calculations showed these interaction were possible for values of d ≤ 3.2 � and 99� ≤ θ ≤ 119� (figure a below). Distances less than 3.2 angstroms, van der Waals surfaces of the C and O on adjacent carbonyls overlap, suggesting that the interaction is quantum mechanical and not classically mediated. Next they calculated d and q values for on high resolution x-ray structures of known proteins. Red shading in the Ramachandran plot in figure c below phi/psi regions in actual proteins that meet the required geometry for np to p* overlap. Hence these geometry are abundant in proteins, especially in secondary structures described above.
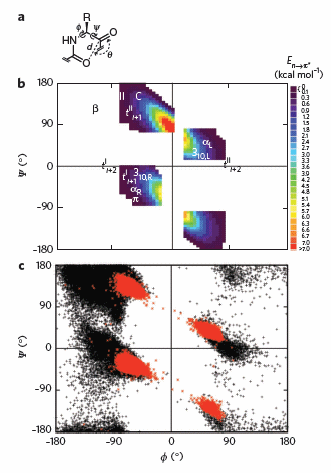
Figure: Ramachandran plots of n to pi* interactions. (a) Definitions of dihedral angles φ (C′i�1�Ni�Cα i�C′i) and ψ (Ni�Cαi�C′i�Ni+1), distance (d) and planar angle θ. Criteria for an n to p* interaction in the crystallographic analyses: d ≤ 3.2 �; 99� ≤ θ ≤ 119�. (b) Computational data showing the energy. Reprinted by permission from Macmillan Publishers Ltd: Nature Chemical Biology. Bartlett, G. et al. n to pi* interactions in protein. 6, pg 615 (2010)