
1.3: Introduction - Water and Buffers
- Page ID
- 7803

Source: BiochemFFA_1_3.pdf. The entire textbook is available for free from the authors at http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy

When it comes to water, we’re literally drowning in it, as water is by far the most abundant component of every cell. To understand life, we begin the discussion with the basics of water, because everything that happens in cells, even reactions buried deep inside enzymes, away from water, is influenced by water’s chemistry.
The water molecule has wide ‘V’ shape (the HO-H angle is 104°) with uneven sharing of electrons between the oxygen and the hydrogen atoms (Figure 1.23). Oxygen, with its higher electronegativity, holds electrons closer to itself than the hydrogens do. The hydrogens, as a result, are described as having a partial positive charge (typically designated as δ+) and the oxygen has a partial negative charge (written as δ- ). Thus, water is a polar molecule because charges are distributed around it unevenly, not symmetrically.
Water as a solvent
Water (Figure 1.23) is described as a solvent because of its ability to solvate (dissolve) many, but not all, molecules. Molecules that are ionic or polar dissolve readily in water, but non-polar substances dissolve poorly in water, if at all. Oil, for example, which is non-polar, separates from water when mixed with it. On the other hand, sodium chloride, which ionizes, and ethanol, which is polar, are able to form hydrogen bonds, so both dissolve in water. Ethanol’s solubility in water is crucial for brewers, winemakers, and distillers – but for this property, there would be no wine, beer or spirits. As explained in an earlier section, we use the term hydrophilic to describe substances that interact well with water and dissolve in it and the term hydrophobic to refer to materials that are non-polar and do not dissolve in water. Table 1.3 illustrates some polar and non-polar substances. A third term, amphiphilic, refers to compounds that have both properties. Soaps, for example are amphiphilic, containing a long, non-polar aliphatic tail and a head that ionizes.

Table 1.3 Image by Aleia Kim
Solubility

The solubility of materials in water is based in free energy changes, as measured by ΔG. Remember, from chemistry, that H is the enthalpy (heat at constant pressure) and S is entropy. Given this,
\[ΔG = ΔH - TΔS\]
where T is the temperature in Kelvin. For a process to be favorable, the ΔG for it must be less than zero.

From the equation, lowered ΔG values will be favored with decreases in enthalpy and/or increases in entropy. Let us first consider why non-polar materials do not dissolve in water. We could imagine a situation where the process of dissolving involves the “surrounding” of each molecule of the nonpolar solute in water, just like each sodium and each chloride ion gets surrounded by water molecules as salt dissolves.
Water organization

There is a significant difference, though between surrounding a non-polar molecule with water molecules and surrounding ions (or polar compounds) with water molecules.
The difference is that since non-polar molecules don’t really interact with water, the water behaves very differently than it does with ions or molecules that form hydrogen bonds. In fact, around each non-polar molecule, water gets very organized, aligning itself regularly. As any freshman chemistry student probably remembers, entropy is a measure of disorder, so when something becomes ordered, entropy decreases, meaning the ΔS is negative, so the TΔS term in the equation is positive (negative of a negative).

Since mixing a non-polar substance with water doesn’t generally have any significant heat component, the ΔG is positive. This means, then, that dissolving a non-polar compound in water is not favorable and does not occur to any significant extent. Further, when the non-polar material associates with itself and not water, then the water molecules are free to mix, without being ordered, resulting in an increase of entropy. Entropy therefore drives the separation of non-polar substances from aqueous solutions.

Amphiphilic substances
Next, we consider mixing of an amphiphilic substance, such as a soap, with water (Figure 1.24). The sodium ions attached to the fatty acids in soap readily come off in aqueous solution, leaving behind a negatively charged molecule at one end and a non-polar region at the other end. The ionization of the soap causes in an increase in entropy - two particles instead of one. The non-polar portion of the negatively charged soap ion is problematic - if exposed to water, it will cause water to organize and result in a decrease of entropy and a positive ΔG.

Since we know fatty acids dissolve in water, there must be something else at play. There is. Just like the non-polar molecules in the first example associated with each other and not water, so too do the non-polar portions of the soap ions associate with each other and exclude water. The result is that the soap ions arrange themselves as micelles (Figure 1.25) with the non-polar portions on the interior of the structure away from water and the polar portions on the outside interacting with water.

The interaction of the polar heads with water returns the water to its more disordered state. This increase in disorder, or entropy, drives the formation of micelles. As will be seen in the discussion of the lipid bilayer, the same forces drive glycerophospholipids and sphingolipids to spontaneously form bilayers where the non-polar portions of the molecules interact with each other to exclude water and the polar portions arrange themselves on the outsides of the bilayer (Figure 1.28).

Yet another example is seen in the folding of globular proteins in the cytoplasm. Nonpolar amino acids are found in the interior portion of the protein (water excluded). Interaction of the non-polar amino acids turns out to be a driving force for the folding of proteins as they are being made in an aqueous solution.
Hydrogen bonds
The importance of hydrogen bonds in biochemistry (Figure 1.30) is hard to overstate. Linus Pauling himself said,
“ . . . . I believe that as the methods of structural chemistry are further applied to physiological problems it will be found that the significance of the hydrogen bond for physiology is greater than that of any other single structural feature.”

In 2011, an IUPAC task group gave an evidence-based definition of hydrogen bonding that states,
“The hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation.”
Partial Charges
The difference in electronegativity between hydrogen and the molecule to which it is covalently bound give rise to partial charges as described above. These tiny charges (δ+ and δ- ) result in formation of hydrogen bonds, which occur when the partial positive charge of a hydrogen atom is attracted to the partial negative of another molecule. In water, that means the hydrogen of one water molecule is attracted to the oxygen of another (Figure 1.31). Since water is an asymmetrical molecule, it means also that the charges are asymmetrical. Such an uneven distribution is what makes a dipole. Dipolar molecules are important for interactions with other dipolar molecules and for dissolving ionic substances (Figure 1.32).
Hydrogen bonds are not exclusive to water. In fact, they are important forces holding together macromolecules that include proteins and nucleic acids. Hydrogen bonds occur within and between macromolecules.

The complementary pairing that occurs between bases in opposite strands of DNA, for example, is based on hydrogen bonds. Each hydrogen bond is relatively weak (compared to a covalent bond, for example - Table 1.4), but collectively they can be quite strong.

Table 1.4 Image by Aleia Kim
Benefits of weak interactions
Their weakness, however, is actually quite beneficial for cells, particularly as regards nucleic acids (Figure 1.33). The strands of DNA, for example, must be separated over short stretches in the processes of replication and the synthesis of RNA. Since only a few base pairs at a time need to be separated, the energy required to do this is small and the enzymes involved in the processes can readily take them apart, as needed. Hydrogen bonds also play roles in binding of substrates to enzymes, catalysis, and protein-protein interaction, as well as other kinds of binding, such as protein-DNA, or antibody-antigen.

As noted, hydrogen bonds are weaker than covalent bonds (Table 1.4) and their strength varies form very weak (1-2 kJ/mol) to fairly strong (29 kJ/mol). Hydrogen bonds only occur over relatively short distances (2.2 to 4.0 Å). The farther apart the hydrogen bond distance is, the weaker the bond is.
The strength of the bond in kJ/mol represents the amount of heat that must be put into the system to break the bond - the larger the number, the greater the strength of the bond. Hydrogen bonds are readily broken using heat. The boiling of water, for example, requires breaking of H-bonds. When a biological structure, such as a protein or a DNA molecule, is stabilized by hydrogen bonds, breaking those bonds destabilizes the structure and can result in denaturation of the substance - loss of structure. It is partly for this reason that most proteins and all DNAs lose their native, or folded, structures when heated to boiling.

Image by Aleia Kim Table 1.5
For DNA molecules, denaturation results in complete separation of the strands from each other. For most proteins, this means loss of their characteristic three-dimensional structure and with it, loss of the function they performed. Though a few proteins can readily reassume their original structure when the solution they are in is cooled, most can’t. This is one of the reasons that we cook our food. Proteins are essential for life, so denaturation of bacterial proteins results in death of any microorganisms contaminating the food.
The importance of buffers
Water can ionize to a slight extent (10-7 M) to form H+ (proton) and OH- (hydroxide). We measure the proton concentration of a solution with pH, which is the negative log of the proton concentration.
pH = -Log[H+]
If the proton concentration, [H+]= 10-7 M, then the pH is 7. We could just as easily measure the hydroxide concentration with the pOH by the parallel equation,
pOH = -Log[OH- ]
In pure water, dissociation of a proton simultaneously creates a hydroxide, so the pOH of pure water is 7, as well. This also means that
pH + pOH = 14
Now, because protons and hydroxides can combine to form water, a large amount of one will cause there to be a small amount of the other. Why is this the case? In simple terms, if I dump 0.1 moles of H+ into a pure water solution, the high proton concentration will react with the relatively small amount of hydroxides to create water, thus reducing hydroxide concentration. Similarly, if I dump excess hydroxide (as NaOH, for example) into pure water, the proton concentration falls for the same reason.
Acids vs bases
Chemists use the term “acid” to refer to a substance which has protons that can dissociate (come off) when dissolved in water. They use the term “base” to refer to a substance that can absorb protons when dissolved in water. Both acids and bases come in strong and weak forms. (Examples of weak acids are shown in Table 1.5.) Strong acids, such as HCl, dissociate completely in water. If we add 0.1 moles (6.02x1022 molecules) of HCl to a solution to make a liter, it will have 0.1 moles of H+ and 0.1 moles of Cl- or 6.02x1022 molecules of each . There will be no remaining HCl when this happens. A strong base like NaOH also dissociates completely into Na+ and OH- .

Weak Acids
Weak acids and bases differ from their strong counterparts. When you put one mole of acetic acid (HAc) into pure water, only a tiny percentage of the HAc molecules dissociate into H+ and Ac- . Clearly, weak acids are very different from strong acids. Weak bases behave similarly, except that they accept protons, rather than donate them. Since we can view everything as a form of a weak acid, we will not use the term weak base here.

Students are often puzzled and expect that [H+] = [A- ] because the dissociation equation shows one of each from HA. This is, in fact, true ONLY when HA is allowed to dissociate in pure water. Usually the HA is placed into solution that has protons and hydroxides to affect things. Those protons and /or hydroxides change the H+ and Aconcentration unequally, since A- can absorb some of the protons and/or HA can release H+ when influenced by the OH- in the solution. Therefore, one must calculate the proton concentration from the pH using the Henderson Hasselbalch equation.
\[pH = pKa + log ([Ac- ]/[HAc])\]

Image by Aleia Kim Table 1.6
You may wonder why we care about weak acids. You may never have thought much of weak acids when you were in General Chemistry. Your instructor described them as buffers and you probably dutifully memorized the fact that “buffers are substances that resist change in pH” without really learning what Clearing Confusion - this meant. Buffers are much too important to be thought of in this way.
UPS
Weak acids are critical for life because their affinity for protons causes them to behave like a UPS. We’re not referring to the UPS that is the United Parcel Service, but instead, to the encased battery backup systems for computers called Uninterruptible Power Supplies that kick on to keep a computer running during a power failure. The battery in a laptop computer is a UPS, for example.
We can think of weak acids as Uninterruptible Proton Suppliers within certain pH ranges, providing (or absorbing) protons as needed. Weak acids thus help to keep the H+ concentration (and thus the pH) of the solution they are in relatively constant.
Consider the bicarbonate/carbonic acid system. Figure 1.35 shows what happens when H2CO3dissociates. Adding hydroxide ions (by adding a strong base like NaOH) to the solution causes the H+ ions to react with OH- ions to make water. Consequently, the concentration of H+ ions would go down and the pH would go up.

However, in contrast to the situation with a solution of pure water, there is a backup source of H+ available in the form of H2CO3. Here is where the UPS function kicks in. As protons are taken away by the added hydroxyl ions (making water), they are partly replaced by protons from the H2CO3. This is why a weak acid is a buffer. It resists changes in pH by releasing protons to compensate for those “used up” in reacting with the hydroxyl ions.

Henderson-Hasselbalch
It is useful to be able to predict the response of the H2CO3 system to changes in H+ concentration. The Henderson-Hasselbalch equation defines the relationship between pH and the ratio of HCO3 - and H2CO3. It is
pH = pKa + log ([HCO3- ]/ [H2CO3])
This simple equation defines the relationship between the pH of a solution and the ratio of HCO3- and H2CO3 in it. The new term, called the pKa, is defined as
pKa = -Log Ka,
just as
pH = -Log [H+].
The Ka is the acid dissociation constant and is a measure of the strength of an acid. For a general acid, HA, which dissociates as
HA ⇄ H+ + A -, Ka = [H+][A- ]/[HA]
Thus, the stronger the acid, the more protons that will dissociate from it when added to water and the larger the value its Ka will have. Large values of Ka translate to lower values of pKa. As a result, the lower the pKa value is for a given acid, the stronger the weak acid is.
Constant pKa
Please note that pKa is a constant for a given acid. The pKa for carbonic acid is 6.37. By comparison, the pKa for formic acid is 3.75. Formic acid is therefore a stronger acid than acetic acid. A stronger acid will have more protons dissociated at a given pH than a weaker acid.
Now, how does this translate into stabilizing pH? Figure 1.35 shows a titration curve. In this curve, the titration begins with the conditions at the lower left (very low pH). At this pH, the H2CO3 form predominates, but as more and more OH- is added (moving to the 45 Why do we care about pH? Because biological molecules can, in some cases, be exquisitely sensitive to changes in it. As the pH of a solution changes, the charges of molecules in the solution can change, as you will see. Changing charges on biological molecules, especially proteins, can drastically affect how they work and even whether they work at all right), the pH goes up, the amount of HCO3- goes up and (correspondingly), the amount of H2CO3 goes down. Notice that the curve “flattens” near the pKa (6.37).
Buffering region
Flattening of the curve tells us is that the pH is not changing much (not going up as fast) as it did earlier when the same amount of hydroxide was added. The system is resisting a change in pH (not stopping the change, but slowing it) in the region of about one pH unit above and one pH unit below the pKa. Thus, the buffering region of the carbonic acid/ bicarbonate buffer is from about 5.37 to 7.37. It is maximally strong at a pH of 6.37.
Now it starts to become apparent how the buffer works. HA can donate protons when extras are needed (such as when OH- is added to the solution by the addition of NaOH). Similarly, A- can accept protons when extra H+ are added to the solution (adding HCl, for example). The maximum ability to donate or accept protons comes when
[A- ] = [HA]
This is consistent with the Henderson Hasselbalch equation and the titration curve. When [A- ] = [HA], pH = 6.37 + Log(1). Since Log(1) = 0, pH = 6.37 = pKa for carbonic acid. Thus for any buffer, the buffer will have maximum strength and display flattening of its titration curve when [A- ] = [HA] and when pH = pKa. If a buffer has more than one pKa (Figure 1.36), then each pKa region will display the behavior.
Buffered vs non-buffered
To understand how well a buffer protects against changes in pH, consider the effect of adding .01 moles of HCl to 1.0 liter of pure water (no volume change) at pH 7, compared to adding it to 1.0 liter of a 1M acetate buffer at pH 4.76. Since HCl completely dissociates, in 0.01M (10-2 M) HCl you will have 0.01M H+. For the pure water, the pH drops from 7.0 down to 2.0 (pH = -log(0.01M)).
By contrast, the acetate buffer’s pH after adding the same amount of HCl is 4.74. Thus, the pure water solution sees its pH fall from 7 to 2 (5 pH units), whereas the buffered solution saw its pH drop from 4.76 to 4.74 (0.02 pH units). Clearly, the buffer minimizes the impact of the added protons compared to the pure water.
Buffer capacity
It is important to note that buffers have capacities limited by their concentration. Let’s imagine that in the previous paragraph, we had added the 0.01 moles HCl to an acetate buffer that had a concentration of 0.01M and equal amounts of Ac- and HAc. When we try to do the math in parallel to the previous calculation, we see that there are 0.01M protons, but only 0.005M A- to absorb them. We could imagine that 0.005M of the protons would be absorbed, but that would still leave 0.005M of protons unbuffered. Thus, the pH of this solution would be approximately
pH = -log(0.005M) = 2.30
Exceeding buffer capacity dropped the pH significantly compared to adding the same amount of protons to a 1M acetate buffer. Consequently, when considering buffers, it is important to recognize that their concentration sets their limits. Another limit is the pH range in which one hopes to control proton concentration.
Multiple ionizable groups
Now, what happens if a molecule has two (or more) ionizable groups? It turns out, not surprisingly, that each group will have its own pKa and, as a consequence, will have multiple regions of buffering.
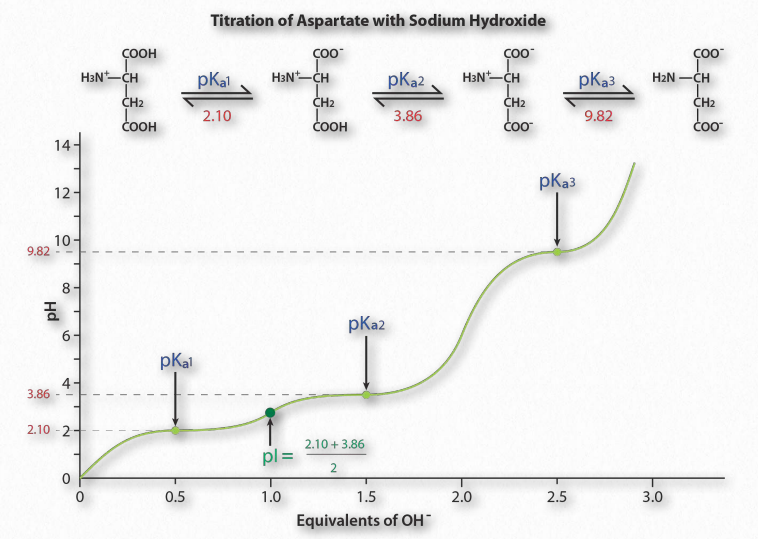
Figure 1.36 shows the titration curve for the amino acid aspartic acid. Note that in- stead of a single flattening of the curve, as was seen for acetic acid, aspartic acid’s titration curve displays three such regions. These are individual buffering regions, each centered on the respective pKa values for the carboxyl group and the amine group.
Aspartic acid has four possible charges: +1 (α-carboxyl group, α-amino group, and Rgroup carboxyl each has a proton), 0 (α- carboxyl group missing proton, α- amino group has a proton, R-group carboxyl has a proton), -1 (α-carboxyl group and R-group carboxyl each lack a proton, α-amino group retains a proton), -2 (α-carboxyl, R-group carboxyl, and α-amino groups all lack extra proton).
Prediction
How does one predict the charge for an amino acid at a given pH? A good rule of thumb for estimating charge is that if the pH is more than one unit below the pKa for a group (carboxyl or amino), the proton is on. If the pH is more than one unit above the pKa for the group, the proton is off. If the pH is NOT more than one or less than one pH unit from the pKa, this simple assumption will not work.
Further, it is important to recognize that these rules of thumb are estimates only. The pI (pH at which the charge of a molecule is zero) is an exact value calculated as the average of the two pKa values on either side of the zero region. It is calculated at the average of the two pKa values around the point where the charge of the molecule is zero. For aspartic acid, this corresponds to pKa1and pKa2.
References
- http://www.lpi.usra.edu/lunar/missions/apollo/ apollo_12/experiments/surveyor/
- Arunan, Elangannan; Desiraju, Gautam R.; Klein, Roger A.; Sadlej, Joanna; Scheiner, Steve; Alkorta, Ibon; Clary, David C.; Crabtree, Robert H.; Dannenberg, Joseph J.; Hobza, Pavel; Kjaergaard, Henrik G.; Legon, Anthony C.; Mennucci, Benedetta; Nesbitt, David J. (2011). "Definition of the hydrogen bond". Pure Appl. Chem. 83 (8): 1637–1641. doi:10.1351/PAC-REC-10-01-02