
19.1: Post-transcriptional Control of Gene Expression
- Page ID
- 42854

Not too long ago we thought that very little of the eukaryotic genome was ever transcribed. We also thought that the only non-coding RNAs were tRNAs and rRNAs. Now we know that other RNAs play roles in gene regulation and the degradation of spent cellular DNA or unwanted foreign DNA. These are discussed in detail below.
A. Ribosomes
The riboswitches is a bacterial transcription mechanism for regulating gene expression. While this mechanism is not specifically post-transcriptional, it is included here because the action occurs after transcription initiation and aborts completion of an mRNA. When the mRNA for an enzyme in the guanine synthesis pathway is transcribed, it folds into stem-&-loop structures. Enzyme synthesis will continue for as long as the cell needs to make guanine. But if guanine accumulates in the cell, excess guanine will bind stem-loop elements near the 5’ end of the mRNA, causing the RNA polymerase and the partially completed mRNA dissociate from the DNA, prematurely ending transcription. The basis of guanine riboswitch regulation of expression of a guanine synthesis pathway enzyme is shown below.

232 Riboswitches Interrupt Bacterial Transcription
The ability to form folded, stem-loop structures at the 5’ ends of bacterial mRNAs seems to have allowed the evolution of translation regulation strategies. Whereas guanine interaction with the stem-loop structure of an emerging 5’ mRNA can abort its own transcription, similar small metabolite/mRNA and even protein/mRNA interactions can also regulate (in this case prevent) translation. As we will see shortly, 5’ mRNA folded structures also play a role in eukaryotic translation regulation.
233 Small Metabolites Also Regulate Bacterial mRNA translation
B. CRISPR/Cas: RNA-Protein Complex of a Prokaryotic Adaptive Immune System
In higher organisms, the immune system is adaptive. It remembers prior exposure to a pathogen, and can thus mount a response to a second exposure to the same pathogen. The discovery of an ‘adaptive immune system’ in many prokaryotes (bacteria, archaebacteria) was therefore something of a surprise.
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) RNAs are derived from phage transcripts that have interacted with CRISPR-Associated (Cas) proteins. They make up the CRISPR/Cas system that seems to have evolved to fight of viral infection by targeting phage DNA for destruction. When viral DNA gets into a cell during a phage infection, it can generate a CRISPR/Cas gene array in the bacterial genome, with spacer DNA sequences separating repeats of the CRISPR genes. These remnants of a phage infection are the memory of this prokaryotic immune system. When a phage attempts to re-infect a previously exposed cell, spacer RNAs and Cas genes are transcribed. After Cas mRNA translation, the Cas protein and spacer RNAs will engage and target the incoming phage DNA for destruction to prevent infection. Thus, the CRISPR/Cas systems (there is more than one!) remember prior phage attacks, and transmit that memory to progeny cells. The CRISPR/Cas9 system in Streptococcus pyogenes is one of the simplest of these immune defense systems (illustrated below).
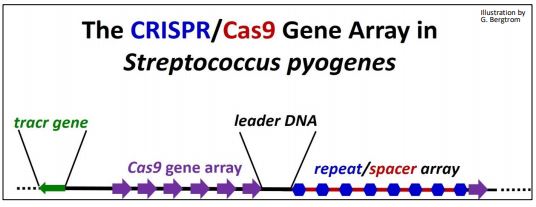
The CRISPR/Cas gene array consists of the following components:
- Cas: Genes native to host cells
- CRISPR: 24-48 bp repeats native to host cells
- Spacer DNA: DNA between CRISPR repeats: typically, phage DNA from prior phage infection or plasmid transformation
- leader DNA: Contains promoter for CRISPR/spacer RNA transcription
- tracr gene: Encodes transcription activator (tracr) RNA (not all systems)
Let's look at CRISPR/Cas in action.
1. The CRISPR/Cas Immune Response
Consider the mechanism of action of this prokaryotic immune system. The action begins when infectious phage DNA gets into the cell, as drawn below.
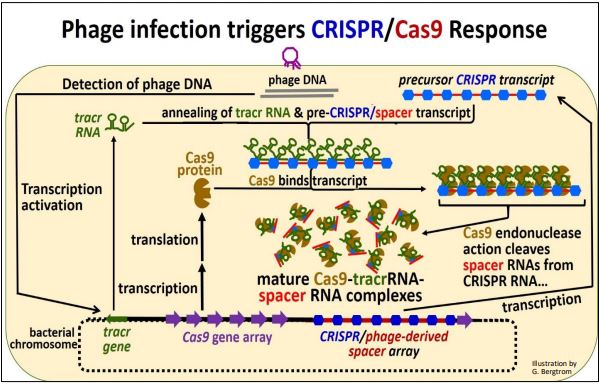
Let’s summarize what has happened here:
a) Incoming phage DNA was detected after phage infection.
b) Then the tracr and Cas genes are transcribed along with the CRISPR/spacer region. Cas mRNAs are translated to make the Cas protein. Remember, the spacer DNAs in the CRISPR region are the legacy of a prior phage infection.
c) CRISPR/spacer RNA forms hydrogen bonds with a complementary region of the tracr RNA as the two RNAs associate with Cas proteins.
d) Cas protein endonucelases hydrolyze spacer RNA from CRISPR RNA sequences. The spacer RNAs remain associated with the complex while the actual, imperfectly palindromic CRISPR sequences (shown in blue in the illustration above) fall off.
In the next steps, phage-derived spacer RNAs, now called guide RNAs (or gRNAs) ‘guide’ mature Cas9/tracrRNA/spacer RNA complexes to new incoming phage DNA resulting from a phage attack. The association of the complex with the incoming phage DNA and subsequent events are illustrated below.
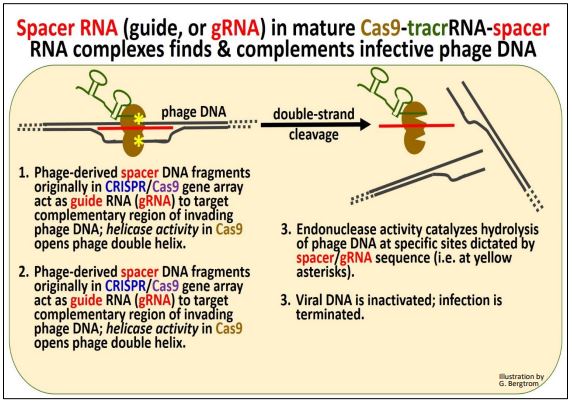
Once again, let’s summarize:
a) Spacer (i.e., gRNA) in the complex targets incoming phage DNA.
b) Cas helicase unwinds incoming phage DNA at complementary regions.
c) gRNA H-bonds to incoming phage DNA.
d) Cas endonucleases create a double-stranded break (hydrolytic cleavage) at specific sites in incoming phage DNA. Because precise site DNA strand cleavage is guided by RNA molecules, CRISPR/Cas endonucleases are classified as type V restriction enzymes.
e) The incoming phage DNA is destroyed and a new phage infection is aborted.
Check out here to learn more about how bacteria acquire spacer DNAs, and therefore how this primitive adaptive immune system ‘remembers’) in the first place
2. Using CRISPR/Cas to Edit/Engineer Genes
Early studies demonstrated the reproducible cleavage of incoming phage DNA at specific nucleotides. Several labs quickly realized that it might be possible to adapt the system to cut DNA at virtually any specific nucleotide in a target DNA! It has turned out that the system works both in vivo and in vitro, allowing virtually unlimited potential for editing genes and RNAs in a test tube… or in any cell. Here is the basic process:
a) Engineer gDNA with a Cas-specific DNA sequence that targets a desired target in genomic DNA.
b) Fuse the gDNA to tracr DNA to make a single guide DNA (sgDNA) so that it can be made as a single guide transcript (sgRNA).
c) Engineer a CRISPR/Cas9 gene array that substitutes this sgDNA for its original spacer DNAs.
d) Place engineered array in a plasmid next to regulated promoters.
e) Transform cells by ‘electroporation’ (works for almost any cell type!)
f) Activate the promoter to transcribe the CRISPR/Cas9 genes…
The applications are powerful… and controversial!
3. The Power and the Controversy
The application of gene editing with CRISPR/Cas systems has already facilitated studies of gene function in vitro, in cells and in whole organisms. Click here for a description of CRISPR/Cas applications already on the market! The efficiency of specific gene editing using CRISPR/Cas systems holds great promise for understanding basic gene structure and function, for determining the genetic basis of disease, and for accelerating the search for gene therapies. Here are just a few examples of how CRISPR/Cas approaches are being applied.
- One can engineer an sgRNA with desired mutations targeting specific sites in chromosomal DNA. Then clone sgRNA into the CRISPR/Cas9 array on a plasmid. After transformation of appropriate cells, the engineered CRISPR/Cas9 forms a complex with target DNA sequences. Following nicking of both strands of the target DNA, DNA repair can insert the mutated guide sequences into the target DNA. The result is loss or acquisition of DNA sequences at specific, exact sites, or Precision Gene Editing. It is the ability to do this in living cells that has excited the basic and clinical research communities.
- Before transforming cells, engineer the CRISPR/Cas9 gene array on the plasmid to eliminate both endonuclease activities from the Cas protein. Upon transcription of the array in transformed cells, the CRISPR/Cas9-sgRNA still finds an sgRNA-targeted gene. However, lacking CAS protein endonuclease activities, the complex that forms just sits there blocking transcription. This technique is sometimes referred to as CRISPRi (CRISPER interference), by analogy to RNAi. Applied to organisms (and not just in vitro or to cells), it mimics the much more difficult knockout mutation experiments that have been used in studies of behavior of cells or organisms rendered unable to express a specific protein.
- There are now several working CRISPR/Cas systems capable of Precision Gene Editing. They are exciting for their speed, precision, their prospects for rapid, targeted gene therapies to fight disease, and their possibilities to alter entire populations (called Gene Drive). By inserting modified genes into the germline cells of target organisms, gene drive can render harmless entire malarial mosquito populations, to eliminate pesticide resistance in e.g. insects, eliminate herbicide resistance in undesirable plants, or genetically eliminate invasive species. For more information, click Gene drive; for an easy read about this process and the controversies surrounding applications of CRISPR technologies to mosquitoes in particular, check out J. Adler, (2016) A World Without Mosquitoes. Smithsonian, 47(3) 36-42, 84.
- It is even possible to delete an entire chromosome from cells. This bit of global genetic engineering relies on identifying multiple unique sequences on a single chromosome and then targeting these sites for CRISPR/Cas. When the system is activated, the chromosome is cut at those sites, fragmenting it beyond the capacity of DNA repair mechanisms to fix the situation. Click here to learn more.
If for no other reason than its efficiency and simplicity, precision gene editing with CRISPR/Cas techniques has raised ethical issues. Clearly, the potential exists for abuse, or even for use with no beneficial purpose at all. It is significant that, as in all discussions of biological ethics, scientists are very much engaged in the conversation. Despite the controversy, we will no doubt continue to edit genes with CRISPR/Cas, and we can look for a near future Nobel Prize for its discovery and application! If you still have qualms, maybe RNA editing will be the answer. Check out the link at Why edit RNA? for an overview of the possibilities!
Finally, “mice and men” (and women and babies too) have antibodies to Cas9 proteins, suggesting prior exposure to microbial CRISPR/Cas9 antigens. This observation may limit clinical applications of the technology! See Uncertain Future of CRISPR-Cas9 Technology.
C. The Small RNAs: miRNA and siRNA in Eukaryotes
Micro RNAs (miRNAs) and small interfering RNAs (siRNAs) are found in C. elegans, a small nematode (roundworm) that quickly became a model for studies of cell and molecular biology and development. The particular attractions C. elegans are that (a) its genome has ~21,700 genes, comparable to the ~25,000 genes in a human genome!; (b) it uses the products of these genes to produce an adult worm consisting of just 1031 cells organized into all of the major organs found in higher organisms; (c) It is possible to trace the embryonic origins of every single cell in its body! C. elegans is illustrated below.
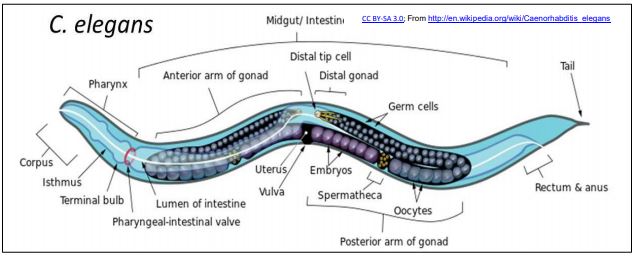
1. Small Interfering RNA (siRNA)
siRNA was first found in plants as well as in C. elegans. However, siRNAs (and miRNAs) are common in many higher organisms. siRNAs were so-named because they interfere with the function of other RNAs foreign to the cell or organism. Their action was dubbed RNA interference (RNAi). For their discovery of siRNAs, A. Z. Fire and C. C. Mello shared the 2006 Nobel Prize in Physiology or Medicine. The action of siRNA targeting foreign DNA is illustrated below.
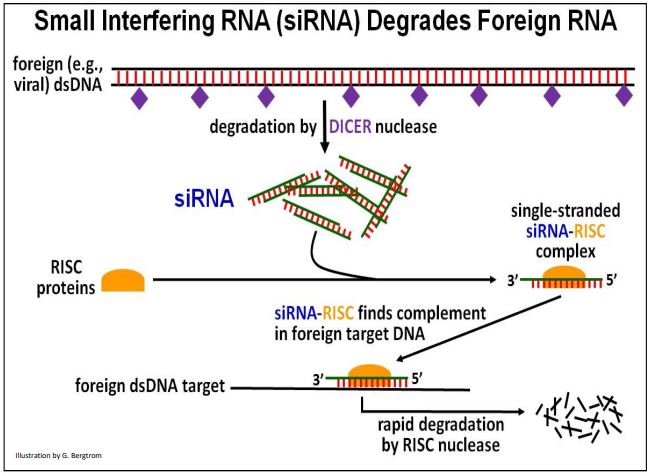
When cells recognize foreign double-stranded RNAs (e.g., some viral RNA genomes) as alien, the DICER a nuclease called hydrolyzes them. The resulting short double-stranded hydrolysis products (the siRNAs) combine with RNAi Induced Silencing Complex, or RISC proteins. The antisense siRNA strand in the resulting siRNA-RISC complex binds to complementary regions of foreign RNAs, targeting them for degradation. Cellular use of RISC to control gene expression in this way may have derived from the use of RISC proteins by miRNAs as part of a cellular defense mechanism, to be discussed next.
Custom-designed siRNAs have been used to disable expression of specific genes in order to study their function in vivo and in vitro. Both siRNAs and miRNAs are being investigated as possible therapeutic tools to interfere with RNAs whose expression leads to cancer or other diseases.
234 siRNA Post Transcriptional Regulation
235 Did siRNA Coopt RISC strategy to Trash Corrupt or Worn out RNA?
For an example check out a Youtube video of unexpected results of an RNAi experiment at this link. In the experiment described, RNAi was used to block embryonic expression of the orthodenticle (odt) gene that is normally required for the growth of horns in a dung beetle. The effect of this knock-out mutation was, as expected, to prevent horn growth. What was unexpected however, was the development of an eye in the middle of the beetle’s head (‘third eye’ in the micrograph).
The 3rd eye not only looks like an eye, but is a functional one. This was demonstrated by preventing normal eye development in odt-knockout mutants. The 3rd eye appeared…, and was responsive to light! Keep in mind that this was a beetle with a 3rd eye, not Drosophila! To quote Justin Kumar from Indiana University, who though not involved in the research, stated that “…lessons learned from Drosophila may not be as generally applicable as I or other Drosophilists, would like to believe … The ability to use RNAi in non-traditional model systems is a huge advance that will probably lead to a more balanced view of development.”
2. Micro RNAs (miRNA)
miRNAs target unwanted endogenous cellular RNAs for degradation. They are transcribed from genes now known to be widely distributed in eukaryotes. The pathway from pre-miRNA transcription through processing and target mRNA degradation is illustrated on the next page.
As they are transcribed, pre-miRNAs fold into a stem-loop structure that is lost during cytoplasmic processing. Like SiRNAs, mature miRNAs combine with RISC proteins. The RISC protein-miRNA complex targets old or no-longer needed mRNAs or mRNAs damaged during transcription.
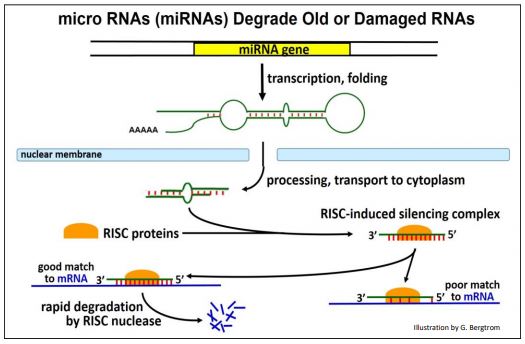
An estimated 250 miRNAs in humans may be sufficient to H-bond to diverse target RNAs; only targets with strong complementarity to a RISC protein-miRNA complex will be degraded.
D. Long Non-Coding RNAs
Long non-coding RNAs (lncRNAs) are a yet another class of eukaryotic RNAs. They include transcripts of antisense, intronic, intergenic, pseudogene and retroposon DNA. Retroposons are one kind of transposon, or mobile DNA element; pseudogenes are recognizable genes with mutations that make them non-functional. While some lncRNAs might turn out to be incidental transcripts that the cell simply destroys, others have a role in regulating gene expression.
A recently discovered lncRNA is XistAR that, along with the Xist gene product, is required to form Barr bodies. Barr bodies form in human females when one of the X chromosomes in somatic cells is inactivated. For a review of lncRNAs, see Lee, J.T. (2012. Epigenetic Regulation by Long Noncoding RNAs; Science 338, 1435-1439).
An even more recent article (at lncRNAs and smORFs) summarizes the discovery that some long non-coding RNAs contain short open reading frames (smORFs) that are actually translated into short peptides of 30+ amino acids! Who knows? The human genome may indeed contain more than 21,000-25,000 protein-coding genes!
E. Circular RNAs (circRNA)
Though discovered more than 20 years ago, circular RNAs (circRNAs) are made in different eukaryotic cell types. Click Circular RNAs (circRNA) to learn more about this peculiar result of alternative splicing. At first circRNAs were hard to isolate. When they were isolated, circRNAs contained “scrambled” exonic sequences and were therefore thought to be nonfunctional errors of mRNA splicing.
In fact, circRNAs are fairly stable. Their levels can rise and fall in patterns suggesting that they are functional molecules. Levels of one circRNA, called circRims1, rise specifically during neural development. In mice, other circRNAs accumulate during synapse formation, likely influencing how these neurons will ultimately develop and function. Thus, circRNAs do not seem to be ‘molecular mistakes’. In fact, errors in their own synthesis may be correlated with disease! Speculation on the functions of circRNAs also includes roles in gene regulation, particularly the genes or mRNAs from which they themselves are derived.
F. "Junk DNA" in Perspective
Not long ago, we thought that less than 5% of a eukaryotic genome was transcribed (i.e., into mRNA, rRNA and tRNA), and that much of the non-transcribed genome served a structural function… or no function at all. The latter, labeled junk DNA, included non-descript intergenic sequences, pseudogenes, ‘dead’ transposons, long stretches of intronic DNA, etc. Thus, junk DNA was DNA we could do without. Junk DNAs were thought to be accidental riders in our genomes, hitchhikers picked up on the evolutionary road.
While miRNA genes are a small proportion of a eukaryotic genome, their discovery, and that of more abundant lnc RNAs suggest a far greater amount of functional DNA in the genome. Might there be in fact, no such thing as “junk DNA”? The debate about how much of our genomic DNA is a relic of past evolutionary experiments and without genetic purpose continues. Read all about it at Junk DNA - not so useless after all and Only 8.2% of human DNA is functional.
Perhaps we need to re-think what it means for DNA to be “junk” or to be without “genetic purpose”. Maintenance of more than 90% of our own DNA with no known genetic purpose surely comes at an energy cost. At the same time, all of that DNA is grist for future selection, a source of the diversity required for long-term survival. The same natural selection that picks up ‘hitchhiker’ DNA sequences, as we have seen, can at some point, put them to work!
G. The RNA Methylome
Call this an RNA epi-transcriptome if you like! Recall that methyl groups direct cleavage of ribosomal RNAs from eukaryotic 45S pre-RNA transcripts. tRNAs among other transcripts, are also post-transcriptionally modified. Known since the 1970s, such modifications were thought to be non-functional. But are they?