
15.3H: Blood Clotting
- Page ID
- 5407

When blood vessels are cut or damaged, the loss of blood from the system must be stopped before shock and possible death occur. This is accomplished by solidification of the blood, a process called coagulation or clotting. A blood clot consists of a plug of platelets enmeshed in a network of insoluble fibrin molecules. Platelet aggregation and fibrin formation both require the proteolytic enzyme thrombin. Clotting also requires calcium ions (Ca2+) (which is why blood banks use a chelating agent to bind the calcium in donated blood so the blood will not clot in the bag) and about a dozen other protein clotting factors. Most of these circulate in the blood as inactive precursors. They are activated by proteolytic cleavage becoming, in turn, active proteases for other factors in the system. By tradition, these factors are designated by Roman numerals.
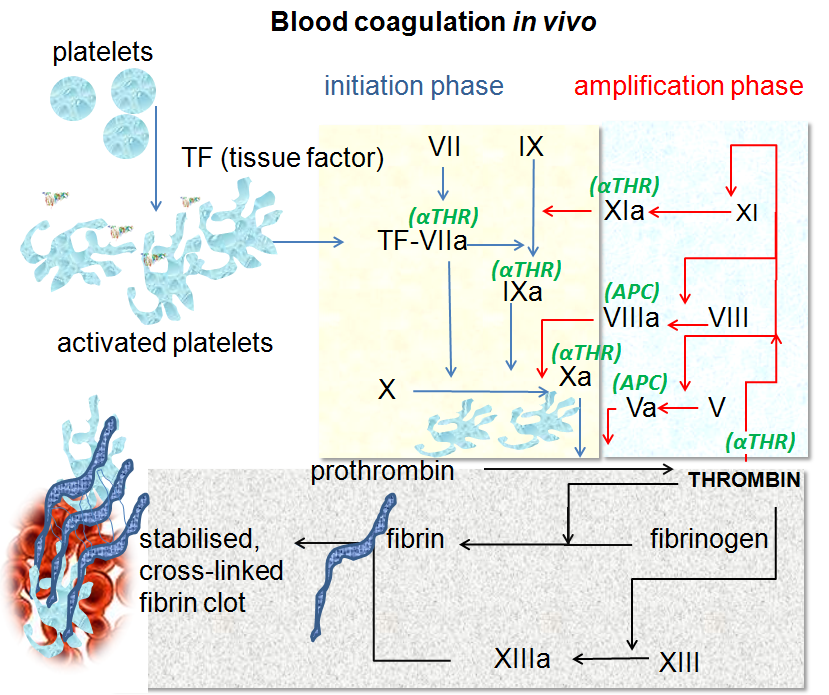
There are two processes that can initiate clotting: A very rapid process the so-called extrinsic pathway or a slower but larger intrinsic pathway.
The Extrinsic Pathway
Damaged cells display a surface protein called tissue factor (TF). Tissue factor then binds to activated Factor 7. The TF-7 heterodimer is a protease with two substrates: Factor 9 and Factor 10.
The Intrinsic Pathway
Factor 12 (also called the Hageman factor) circulates in the blood. If blood escapes into tissue spaces (e.g., as a result of an injury), contact with collagens in the tissue space activates Factor 12. Activated Factor 12 is a serine protease that activates Factor 11 which in turn activates Factor 9 which in turn activates Factor 10.
The Two Pathways Converge at Factor 10
Factor 10 - produced by either or, more likely, both pathways - binds and activates Factor 5. This heterodimer is called prothrombinase because it is a protease that converts prothrombin (also known as Factor II) to thrombin. Thrombin has several different activities. Two of them are:
- proteolytic cleavage of fibrinogen (aka "Factor I") to form soluble molecules of fibrin and a collection of small and fibrinopeptides.
- activation of Factor 13 which forms covalent bonds between the soluble fibrin molecules converting them into an insoluble meshwork — the clot.
Thrombin and activated Factors 10 ("Xa") and 11 ("XIa") are also serine proteases.
Amplifying the Clotting Process
The clotting process also has several positive feedback loops which quickly magnify a tiny initial event into what may well be a lifesaving plug to stop bleeding.
- The TF-7 complex (which started the process) also activates Factor 9.
- Factor 9 binds to Factor 8, a protein that circulates in the blood stabilized by another protein, von Willebrand Factor (vWF).
- This complex activates more Factor 10.
- As thrombin is generated, it activates more
- Factor 5
- Factor 8
- Factor 11 (all shown above with green arrows).
- Factor 11 amplifies the production of activated Factor 9.
Thus what may have begun as a tiny, localized event rapidly expands into a cascade of activity.
Platelets
Platelets are cell fragments produced from megakaryocytes. Blood normally contains 150,000 to 400,000 per microliter (µl). If this value should drop much below 20,000/µl, there is a danger of uncontrolled bleeding. This is because of the essential role of platelets in maintaining the integrity of the adherens junctions that provide a tight seal between the endothelial cells that line the blood vessels and in forming a clot where blood vessels have been broken.
When blood vessels are damaged, fibrils of collagen in the extracellular matrix (ECM) are exposed. Platelets then begin to adhere to the collagen through the action of specific receptors for collagen present on their plasma membrane and von Willebrand factor which links the platelets to the collagen. These actions cause a plug of platelets to form at the site.
The bound platelets release ADP and thromboxane A2, which recruit and activate still more platelets circulating in the blood. (This role of thromboxane accounts for the beneficial effect of low doses of aspirin — a cyclooxygenase inhibitor — in avoiding heart attacks.), tissue factor, and serotonin, which enhances their clumping and promotes constriction of the blood vessel.
ReoPro s a monoclonal antibody directed against platelet receptors. It inhibits platelet aggregation and appears to reduce the risk that "reamed out" coronary arteries (after coronary angioplasty) will plug up again.
Bleeding Disorders
A deficiency of a clotting factor can lead to uncontrolled bleeding. The deficiency may arise because not enough of the factor is produced or a mutant version of the factor fails to perform properly.
Examples:
- von Willebrand disease (the most common)
- hemophilia A for factor 8 deficiency
- hemophilia B for factor 9 deficiency.
- hemophilia C for factor 11 deficiency
In some cases of von Willebrand disease, either a deficient level or a mutant version of the factor eliminates its protective effect on factor 8. The resulting low level of factor 8 mimics hemophilia A.
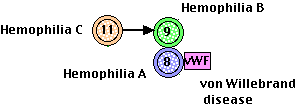
Why do all the human bleeding disorders involve factors in amplification pathways? Probably because they are the only deficiencies that can be tolerated. Loss of the genes for tissue factor or factor 7 in knockout mice is lethal.
Hemophilia A and B
The genes encoding factors 8 and 9 are on the X chromosome. Thus their inheritance is X-linked. Like other X-linked disorders, hemophilia A and B are found almost exclusively in males because they inherit just a single X chromosome, and if the gene for factor 8 (or 9) on it is defective, they will suffer from the disease.
Queen Victoria of the UK was a carrier of a mutant factor 9 gene and passed it on to several of her descendants.
There are many different mutant versions of the genes for factors 8 and 9. Although some produce only a minor effect on the function of their protein, others fail to produce any functioning clotting factor.
Treating Hemophilia A and B
What can be done?
Factor 8 and 9 can be extracted from donated blood, usually pooled from several thousand donors, and purified. Injections of this material can halt episodes of bleeding in hemophiliacs and have allowed countless young men to live relatively normal lives. However, in the early 1980s, blood contaminated with the human immunodeficiency virus (HIV) was unknowingly used to manufacture preparations of factors 8 and 9. In some areas, 90% or more of the hemophiliacs became infected by these contaminated preparations. Many have since died of AIDS. The future now looks brighter because:
- all donated blood is now tested to see if the donor has been infected with HIV (as well as hepatitis B and C)
- plasma-derived preparations of factors 8 and 9 are now treated with heat and/or solvents to destroy any viruses that might be present
- recombinant factor 8 and recombinant factor 9 made by genetic engineering are now available
These recombinant factors are made by inserting the DNA encoding the human protein into mammalian cells grown in culture. E. coli cannot be used because these factors are glycoproteins, and E. coli lacks the machinery to attach carbohydrate properly.
And the team that brought us Dolly reported in the 19 December 1997 issue of Science that they have succeeded in cloning female sheep transgenic for the human factor 9 gene. The human gene is coupled to the promoter for the ovine (sheep) milk protein beta-lactoglobulin. When the lambs mature, it is hoped that they will secrete large amounts of human factor 9 in their milk, which can then be purified for human therapy.
Attempts to cure hemophilia by gene therapy also look promising. It is difficult to see how even the most worried critics of genetic engineering can fail to approve its potential to save the lives of thousands of hemophiliacs in the years to come.
Liver Transplants
People with liver failure can be cured with a liver transplant. On the rare occasions when the patient has happened to be a hemophiliac (A, B or C), the transplant cured not only the patient's liver disease but cured his hemophilia as well!
Controlling Clotting
While the ability to clot is essential to life, the process must be carefully regulated. Inappropriate clot formation, especially in the brain or lungs, can be life-threatening.
Antithrombin III
As its name suggests, this plasma protein (a serpin) inhibits the formation of thrombin. It does so by binding to and thus inactivating prothrombin, factor 9 and factor 10. Heparin is a mixture of polysaccharides that bind to antithrombin III, inducing an allosteric change that greatly enhances its inhibition of thrombin synthesis. Some surgical patients, especially those receiving hip or heart valve replacements, and people at risk of ischemic stroke (clots in the brain), are given heparin.
Protein C
With its many clot-promoting activities, it is probably no accident that thrombin sits at the center of the control mechanism.
- Excess thrombin binds to cell-surface receptors called thrombomodulin.
- The resulting complex activates a plasma protein called Protein C and its cofactor Protein S.
- Together these inhibit further thrombin formation
- directly — by inactivating Factor 5
- indirectly — by inactivating Factor 8.
Some inherited disorders that predispose to spontaneous clots, especially in the leg veins:
- inherited deficiency of Protein C or Protein S
- inherited mutation in the Factor 5 gene producing a protein that no longer responds to the inhibitory effect of Protein C
Recombinant Protein C is now available to treat people threatened with inappropriate clotting, e.g., as a result of widespread infection (sepsis).
Vitamin K
Vitamin K is a cofactor needed for the synthesis (in the liver) of factors 2 (prothrombin), 7, 9, and 10 and proteins C and S. A deficiency of Vitamin K predisposes to bleeding. Conversely, blocking the action of vitamin K helps to prevent inappropriate clotting.
Warfarin (Coumadin®) is sometimes prescribed as a "blood thinner" because it is an effective vitamin K antagonist. (Warfarin is also used as a rat poison because it can cause lethal internal bleeding.) Warfarin treatment is tricky because the therapeutic window (neither too much nor too little) is very narrow, and there is substantial variability between people in their response. So treatment requires regular monitoring of clotting time until the proper dosage is established. However, a number of new anti-clotting agents — that work by inhibiting activated factor 10 (Factor Xa) — are being tested and may turn out to be safer and effective alternatives to warfarin.
Dissolving clots
Plasma contains plasminogen, which binds to the fibrin molecules in a clot. Nearby healthy cells release tissue plasminogen activator (TPA), which also binds to fibrin and, as its name suggests, activates plasminogen forming plasmin. Plasmin (another serine protease) proceeds to digest fibrin, thus dissolving the clot.
Recombinant human TPA is now produced by recombinant DNA technology. Injected within the first hours after a heart attack, it has saved many lives by dissolving the clot blocking the coronary artery and restoring blood flow before the heart muscle becomes irreversibly damaged. It is also used for people who suffer an ischemic stroke; that is, a clot in the brain. (It must not, of course, be used for hemorrhagic strokes, that is, a burst blood vessel)
Angiogenesis
Thrombin (as well as factors 7 and 10) promotes healing by stimulating the growth of new blood vessels at the site of damage.