
20: Transcriptional regulation via chromatin alterations
- Page ID
- 391

REGULATION BY CHANGES IN CHROMATIN STRUCTURE
Review of nucleosome and chromatin structure
Nucleosome composition
- Nucleosomes are the repeating subunit of chromatin.
- Nucleosomes are composed of a nucleosome core, histone H1 (in higher eukaryotes) and variable length linker DNA (0-50bp).
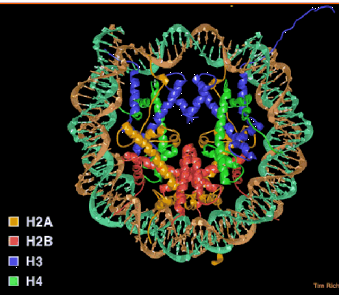
- The nucleosome core contains an octamer of 2 each of the core histones (H2A, H2B, H3 and H4) and 146 bp of DNA wrapped 1.75 turns (Figure 4.6.1).
Histone interactions in the nucleosome
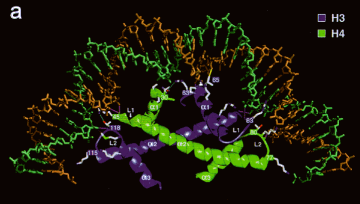
- Core histones dimerize through their histone fold motifs generating H3/H4 dimers and H2A H2B dimers (Figure 4.6.2.).
- Two H3/H4 dimers associate to form a tetramer, which binds DNA.
- Two H2A/H2B dimers associate with the tetramer to form the histone octamer.
- At physiological salt the octamer is not stable unless bound to DNA and dissociates into the H3/H4 tetramer and two H2A/H2B dimers.
Chromatin higher order structure
- Arrays of nucleosomes condense into higher order chromatin fibers (Figure 4.6.3.).
- Despite over 2 decades of investigation the structure of the “30nm” chromatin fiber is not known.
- This may be due to irregularity or instability of the structure.
- This level of structure has been implicated in mechanisms of chromatin repression; thus, the lack of structural information at this level is particularly troublesome

Biochemical investigation of different states of chromatin and gene activity in cells
Sensitivity of chromatin to nucleases
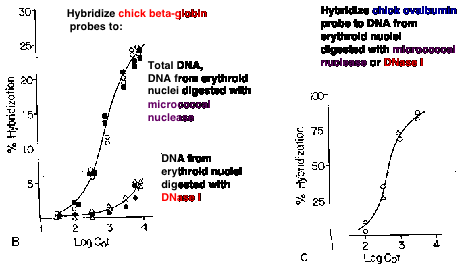
Proposed sequence for gene activation
1. Open a chromatin domain
Relocate away from pericentromeric heterochromatin
Establish a locus-wide open chromatin configuration
General histone hyperacetylation
DNase I sensitivity
2. Activate transcription
Local hyperacetylation of histone H3
Promoter activation to initiate and elongate transcription
Summary of cis-regulatory elements that act in chromatin
Generate an open, accessible chromatin structure
Can extend over about hundreds of kb
Can be tissue specific
Enhance expression of individual genes
Can be tissue specific
Can function at specific stages of development.
Insulate genes from position effects.
Enhancer blocking assay
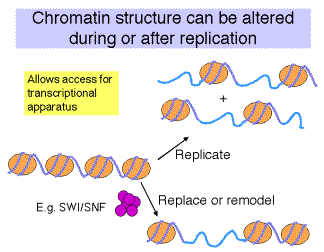
How is the structure of chromatin modified in cells to change transcriptional activity?
Competition vs. Replacement models for how transcription factors occupy their binding sites on a chromatin template.
The HAT complexes could be involved in other processes, or can affect them indirectly through their effects on transcription. For instance, one component of the SAGA HAT complex is Tra1, the yeast homolog of a human protein involved in cellular transformation. It may be a direct target of activator proteins.
Multiple nuclear HATs are found in yeast and in other species (Table 4.6.2). They are all large with many subunits. By comparison, their substrate, which is the nucleosome, is 0.2 MDa in mass. They have different substrate specificities. Some acetylated H3 preferentially, others acetylate H4. The reason for the diversity of HATs is a matter of current study.
Table 4.6.2.The four major nuclear HAT complexes in yeast
| Complex | Mass (MDa=megadaltons) | |
| SAGA | 1.8 | |
| NuA4 | 1.4 | |
| ADA | 0.8 | |
| NuA3 | 0.5 |