
1: Fundamental Properties of Genes
- Page ID
- 292


Gene mapping by conjugal transfer
Conjugal transfer can also be used for genetic mapping. By using many different hfr strains, each with the F factor integrated at a different part of the E. colichromosome, the positions of many genes were mapped. These studies showed that the genetic map of the E. coli chromosome is circular. During conjugal transfer, genes closer to the site of F integration are transferred first. By disrupting the mating at different times, one can determine which genes are closer to the integration site. Thus on the E. colichromosome, genes are mapped in terms of minutes (i.e., the time it takes to transfer to recipient).
For example, for an hfr strain with the F factor integrated at 0 min on the E. colimap, conjugal transfer to a female recipient would transfer
- leuACBD at 1.7 min
- pyrH at 4.6 min
- proAB at 5.9 min
- bioABFCD at 17.5 min.

Bacteriophage
Bacteriophageare viruses that infect bacteria. Because of their very large number of progeny and ability to recombine in mixed infections (more than one strain of bacteria in an infection), they have been used extensively in high-resolution definition of genes. Much of what we know about genetic fine structure, prior to the advent of techniques for isolating and sequencing genes, derive form studies in bacteriophage.
Bacteriophage have been a powerful model genetic system, because they have small genomes, have a short life cycle, and produce many progeny from an infected cell. They provide a very efficient means for transfer of DNA into or between cells. The large number of progeny makes it possible to measure very rare recombination events.
Lytic bacteriophage form plaqueson lawns of bacteria; these are regions of clearing where infected bacteria have lysed. Early work focused on mutants with different plaque morphology, e.g. T2 r, which shows rapid lysis and generates larger plaques, or on mutants with different host range, e.g. T2 h, which will kill both host strains B and B/2.
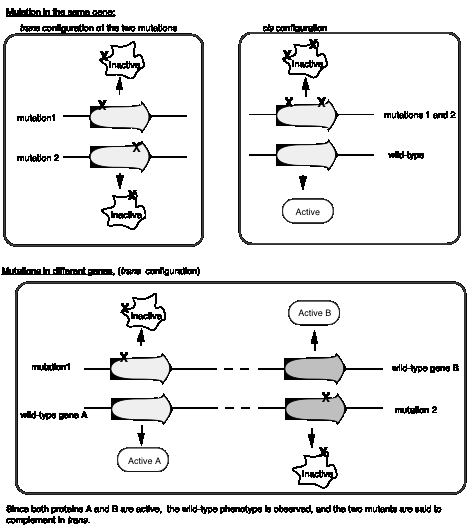
A cis-trans complementation test defines a cistron, which is a gene
Seymour Benzer used the rIIlocus of phage T4 to define genes by virtue of their behavior in a complementation test, and also to provide fundamental insight into the structure of genes (in particular, the arrangement of mutable sites - see the next section). The difference in plaque morphology between rand r+phage is easy to see (large versus small, respectively), and Benzer isolated many r mutants of phage T4. The wild type, but not any rIImutants, will grow on E. colistrain K12(l), whereas both wild type and mutant phage grow equally well on E. colistrain B. Thus the wild phenotype is readily detected by its ability to grow in strain K12 (l).
If E. colistrain K12 (l) is co-infected with 2 phage carrying mutations at different positions in rIIA, you get no multiplication of the phage (except the extremely rare wild type recombinants, which occur at about 1 in 106 progeny). In the diagram below, each line represents the chromosome from one of the parental phage.
rIIA rIIB
phage 1 _|__x______|________|_
phage 2 _|_______x_|________|_
Likewise, if the two phage in the co-infection carry mutations at different positions in rIIB, you get no multiplication of the phage (except the extremely rare wild type recombinants, about 1 in 106).
rIIA rIIB
phage 3 _|_________|_x______|_
phage 4 _|_________|______x_|_
However, if one of the co-infecting phage carries a mutation in rIIAand the other a mutation in rIIB, then you see multiplication of the phage, forming a very large number of plaques on E. colistrain K12 (l).
rIIA rIIB
phage 1 _|__x______|________|_ Provides wt rIIB protein
phage 4 _|_________|______x_|_ Provides wt rIIA protein
Together these two phage provide all the phage functions - they complementeach other. This is a positive complementation test. The first two examples show no complementation, and we place them in the same complementation group. Mutants that do not complement are placed in the same complementation group; they are different mutant alleles of the same gene. Benzer showed that there were two complementation groups (and therefore two genes) at the r II locus, which he called A and B.
In the mixed infection with phage 1 and phage 4, you also obtain the rare wild type recombinants, but there are more recombinants than are seen in the co-infections with different mutant alleles. Why?
Consider the results of infection of a bacterial culture with two mutant alleles of gene rIIA.
T4rIIA6 _|_______________________x______|_
and T4rIIA27 _|_______x______________________|_
(x marks the position of the mutation in each allele).
Progeny phage from this infection include those with a parental genotype (in the great majority), and at a much lower frequency, two types of recombinants:
wild type T4 r+ _|______________________________|_
double mutant T4rIIA6 rIIA27 _|_______x_______________x______|_
The wild type is easily scored because it, and not any rIImutants, will grow on E. coli strain K12(l), whereas both wild type and mutant phage grow equally well on E. coli strain B. Thus you can selectfor the wild type (and you will see only the desired recombinant). Finding the double mutants is more laborious, because they are obtained only by screening through the progeny, testing for phage that when backcrossed with the parental phage result in no wild type recombinant progeny.
Equal numbers of wild type and double mutant recombinants were obtained, showing that recombination can occur within a gene, and that this occurs by reciprocal crossing over. If recombination were only between genes, then no wild type phage would result. A large spectrum of recombination values was obtained in crosses for different alleles, just like you obtain for crosses between mutants in separate genes.
Several major conclusions could be made as a result of these experiments on recombination within the rIIgenes.
- A large number of mutable sites occur within a gene, exceeding some 500 for the rIIA and rIIBgenes. We now realize that these correspond to the individual base pairs within the gene.
- The genetic maps are clearly linear, indicating that the gene is linear. Now we know a gene is a linear polymer of nucleotides.
- Most mutations are changes at one mutable site (point mutations). Many genes can be restored to wild type by undergoing a reverse mutation at the same site (reversion).
- Other mutations cause the deletionof one or more mutable sites, reflecting a physical loss of part of the rII gene. Deletions of one or more mutable site (base pair) are extremely unlikely to revert back to the original wild type.
One gene encodes one polypeptide
One of the fundamental insights into how genes function is that one gene encodes one enzyme (or more precisely, one polypeptide). Beadle and Tatum reached this conclusion based on their complementation analysis of the genes required for arginine biosynthesis in fungi. They showed that a mutation in each gene led to a loss of activity of one enzyme in the multistep pathway of arginine biosynthesis. As discussed above in the section on genetic dissection, a large number of Arg auxotrophs (requiring Arg for growth) were isolated, and then organized into a set of complementation groups, where each complementation groups represents a gene.
The classic work of Beadle and Tatum demonstrated a direct relationship between the genes defined by the auxotrophic mutants and the enzymes required for Arg biosynthesis. They showed that a mutation in one gene resulted in the loss of one particular enzymatic activity, e.g. in the generalized scheme below, a mutation in gene 2 led to a loss of activity of enzyme 2. This led to an accumulation of the substrate for that reaction (intermediate N in the diagram below). If there were 4 complementation groups for the Arg auxotrophs, i.e. 4 genes, then 4 enzymes were found in the pathway for Arg biosynthesis. Each enzyme was affected by mutations in one of the complementation groups.
Intermediates:
M ® N ® O ® P ® Arg
enzyme 1 enzyme 2 enzyme 3 enzyme 4
gene 1 gene 2 gene 3 gene 4
Figure 1.15. A general scheme showing the relationships among metabolic intermediates (M, N, O, P), and end product (Arg), enzymes and the genes that encode them.
In general, each step in a metabolic pathway is catalyzed by an enzyme (identified biochemically) that is the product of a particular gene (identified by mutants unable to synthesize the end product, or unable to break down the starting compound, of a pathway). The number of genes that can generate auxotrophic mutants is (usually) the same as the number of enzymatic steps in the pathway. Auxotrophic mutants in a given gene are missing the corresponding enzyme. Thus Beadle and Tatum concluded that one gene encodes one enzyme. Sometimes more than one gene is required to encode an enzyme because the enzyme has multiple, different polypeptide subunits. Thus each polypeptide is encoded by a gene.
The metabolic intermediates that accumulate in each mutant can be used to place the enzymes in their order of actionin a pathway. In the diagram in Figure 1.15, mutants in gene 3 accumulated substance O. Feeding substance O to mutants in gene 1 or in gene 2 allows growth in the absence of Arg. We conclude that the defects in enzyme 1 or enzyme 2, respectively, are upstream of enzyme 3. In contrast, feeding substance O to mutants in gene 4 will not allow growth in the absence of Arg. Even though this mutant can convert substance O to substance P, it does not have an active enzyme 4 to convert P to Arg. The inability of mutants in gene 4 to grow on substance O shows that enzyme 4 is downstream of enzyme 3.
Imagine that you are studying serine biosynthesis in a fungus. You isolate serine auxotrophs, do all the pairwise crosses of the mutants and discover that the auxotrophs can be grouped into three complementation groups, called A, B and C. You also discover that a different metabolic intermediate accumulates in members of each complementation group - substance A in auxotrophs in the A complementation group, substance B in the B complementation group and substance C in the C complementation group. Each of the intermediates is fed to auxotrophs from each of the three complementation groups as tabulated below. A + means that the auxotroph was able to grow in media in the absence of serine when fed the indicated substance; a - denotes no growth in the absence of serine.
| Fed: | mutant in complementation group A | mutant in complementation group B | mutant in complementation group C |
|---|---|---|---|
| substance A | - | + | + |
| substance B | - | - | - |
| substance C | - | + | - |
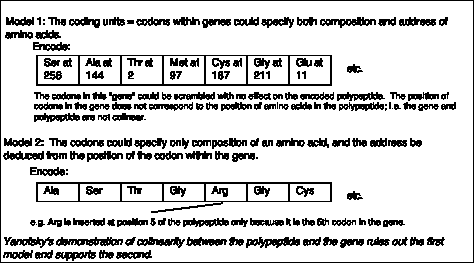
Eukaryotic mRNAs have covalent attachment of nucleotides at the 5' and 3' ends, and in some cases nucleotides are added internally (a process called RNA editing). Recent work shows that additional nucleotides are added post‑transcriptionally to some bacterial mRNAs as well.
Regulatory signals can be considered parts of genes
In order to express a gene at the correct time, the DNA also carries signals to start transcription (e.g. promoters), signals for regulating the efficiency of starting transcription (e.g. operators, enhancers or silencers), and signals to stop transcription (e.g. terminators). Minimally, a gene includes the transcription unit, which is the segment of DNA that is copied into RNA in the primary transcript. The signals directing RNA polymerase to start at the correct site, and other DNA segments that influence the efficiency of this process are regulatory elements for the gene. One can also consider them to be part of the gene, along with the transcription unit.
A contemporary problem - finding the function of genes
Genes were originally detected by the heritable phenotype generated by their mutant alleles, such as the white eyes in the normally red-eyed Drosophilaor the sickle cell form of hemoglobin (HbS) in humans. Now that we have the ability to isolate virtually any, and perhaps all, segments of DNA from the genome of an organism, the issue arises as to which of those segments are genes, and what is the function of those genes. (The genomeis all the DNA in the chromosomes of an organism.) Earlier geneticists knew what the function of the genes were that they were studying (at least in terms of some macroscopic phenotype), even when they had no idea what the nature of the genetic material was. Now molecular biologists are confronted with the opposite problem - we can find and study lots of DNA, but which regions are functions? Many computational approaches are being developed to guide in this analysis, but eventually we come back to that classical definition, i.e. that appropriate mutations in any functional gene should generate a detectable phenotype. The approach of biochemically making mutations in DNA in the laboratory and then testing for the effects in living cells or whole organisms is called "reverse genetics."
Additional Readings
- Griffiths, A. J. F., Miller, J. H., Suzuki, D. T., Lewontin, R. C. and Gelbart, W. M. (1993) An Introduction to Genetic Analysis, Fifth Edition (W. H. Freeman and Company, New York).
- Cairns, J., Stent, G. S. and Watson, J. D., editors (1992) Phage and the Origins of Molecular Biology, Expanded Edition (Cold Spring Harbor Laboratory Press, Plainview, NY).
- Brock, T. D. (1990) The Emergence of Bacterial Genetics (Cold Spring Harbor Laboratory Press, Plainview, NY).
- Benzer, S. (1955) Fine structure of a genetic region in bacteriophage. Proceedings of the National Academy of Sciences, USA 47: 344-354.
- Yanofsky, C. (1963) Amino acid replacements associated with mutation and recombination in the A gene and their relationship to in vitro coding data. Cold Spring Harbor Symposia on Quantitative Biology 18: 133-134.
- Crick, F. (1970) Central dogma of molecular biology. Nature 227:561-563
Questions
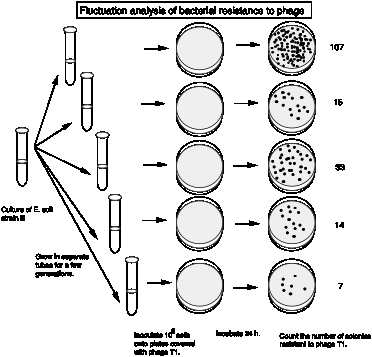
The actual results from Luria and Delbrück are summarized in the following table. They examined 87 cultures, each with 0.2 ml of bacteria, for phage resistant colonies.
| Number of resistant bacteria | Number of cultures |
| 0 | 29 |
| 1 | 17 |
| 2 | 4 |
| 3 | 3 |
| 4 | 3 |
| 5 | 2 |
| 6-10 | 5 |
| 11-20 | 6 |
| 21-50 | 7 |
| 51-100 | 5 |
| 101-200 | 2 |
| 201-500 | 4 |
| 501-1000 | 0 |
Interested students may wish to read about the re-examination of the origin of mutations by Cairns, Overbaugh and Miller (1988, The origin of mutants. Nature 335:142-145). Using a non-lethal selective agent (lactose), they obtained results indicating both pre-adaptive (spontaneous) mutations as well as some apparently induced by the selective agent.