
9.5: Threshold Models
- Page ID
- 21632

Recently, Joe Felsenstein (2005, 2012) introduced a model from quantitative genetics, the threshold model, to comparative methods. Threshold models work by modeling a discrete character as underlain by some other, unobserved, continuous trait (called the liability). If the liability crosses a certain threshold value, then the discrete state changes. More specifically, we can consider a single trait, y, with two states, 0 and 1, which is in turn determined by some underlying continuous variable, x, called the liability. If x is greater than the threshold, t, then y is 1; otherwise, y is 0. Felsenstein (2005) assumes that x evolves under a Brownian motion model, although other models like OU are, in principle, possible.
We can find the likelihood to this model by considering the observations of character states at the tips of the tree. We observe the state of each species, yi. We do not know the liability values for these species. However, we treat these liabilities as unobserved and consider their distributions. Under a Brownian motion model, we know that the liabilities will follow a multivariate normal distribution (see chapter 3). We can calculate the probability of observing the data (yi) by finding the integral of the distributions of liabilities on the side of the threshold that matches the data. So if the distribution of the liability for species i is pi(x), then:
\[ p(y_i = 0) = {\int\limits_{-\infty}^{t} p_i (x) dx} \label{9.1}\]
and
\[ p(y_i = 1) = {\int\limits_{t}^{\infty} p_i (x) dx} \]
(see Figure 9.3 for an illustration of this calculation, which is easier than it looks since there are standard formulas for finding the area under a normal distribution).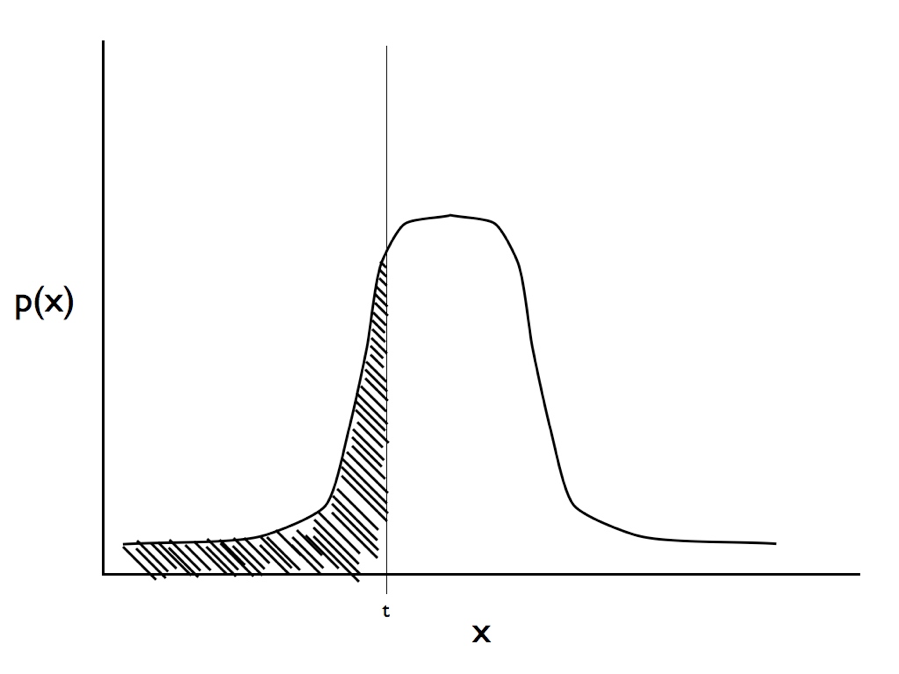
Figure 9.3. Illustration of the integral in Equation \ref{9.1}. For a trait with observed state zero we calculate the area under the curve from negative infinity to the threshold t. Image by the author, can be reused under a CC-BY-4.0 license.
One can fit this model using standard ML or Bayesian methods. Current implementations include an expectation-maximization (EM) algorithm (Felsenstein 2005, 2012) and a Bayesian MCMC (Revell 2014).
The threshold model differs in some key ways from standard Mk-type models. First of all, threshold characters evolve differently than non-threshold characters because of their underlying liability. In particular, the effective rate of change of the discrete character depends on the amount of time that a lineage has been in that character state. Characters that have just changed (say, from 0 to 1) are likely to change back (from 1 to 0), since the liability is likely to be near the threshold. By contrast, characters that have been in one state or the other for a long time tend to be more unlikely to change (since the liability is likely very far from the threshold). This difference matches biological intuition for some characters, where millions of years in one state means that change to a different state might be unlikely. This behavior of the threshold model can potentially account for variation in transition rates across clades without adding additional model parameters. Second, the threshold model scales to cover more than one character more readily than Mk models. Finally, in a threshold framework, it is straightforward to extend the model to include a mixture of both discrete and continuous characters – basically, one assumes that the continuous characters are like “observed liabilities,” and can be modeled together with the discrete characters.