
27.6: Reconstruction
- Page ID
- 41072
In the previous section we learned how to compare and combine gene trees and species trees. In this section, we will use this information to reconstruct gene trees and species trees.
Species Tree Reconstruction
In the past, it was really hard to identify a marker gene that would give insight into the differentiation for a specific species. As sequencing improved, we started having lots of sequencing data on various genes. Based on different sets of loci, people built different trees, which were highly dependent on the set of loci chosen. Possible reasons why trees differ include noise (from statistical estimate errors and noise), hidden duplications and losses, and allele sorting in a population.
Species Tree Reconstruction Problem
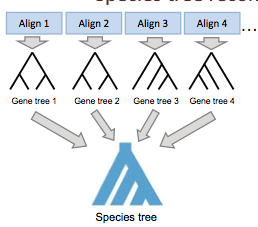
Given lots of different gene trees that disagree, our goal is to make them into one species tree (as shown in Figure 27.13. There are lots of different algorithms that reconstruct species trees. These algorithms include Supermatrix methods (Rokas 2003, Ciccareli 2006), Supertree methods (Creevey & McInerney 2005), Minimizing Deep Coalescence (Maddison & Knowles 2006) and Modeling coalescence (Liu & Pearl 2007).
One way to do this, which is mostly effective for noisy data, is to pull more data together in order to increase accuracy. This is done by concatenating gene alignments into a super-matrix.
Another method involves building a tree for each one and using a consensus method to summarize these trees. Then we identify analogous branches across the a lot of trees and build a species tree that has the branches that occur most frequently.
There is another way to reconstruct a species tree, which is effective in case the gene trees disagree because of duplications and losses. The goal is to find the species tree that applies the fewest duplications. We build all the gene trees and then propose a species tree. Next, we use reconciliation to determine the number of events each gene tree combined with the proposed species tree implies. Then, we propose other species trees and move branches around. Wrong species trees tend to have lots of events that did not happen. The correct tree should have the fewest number of events.
Improving Gene Tree Reconstruction and Learning Across Gene Trees
We can use methods similar to those described above to build better gene trees. This can be done by using information from a species tree to study a gene tree of interest. For example, species trees can be used to determine when losses and duplications occurred. The idea is that we can use the fact that species trees are often built from the entire genome, to obtain more information about related gene trees. We can use both the branch length and the number of events to do this.
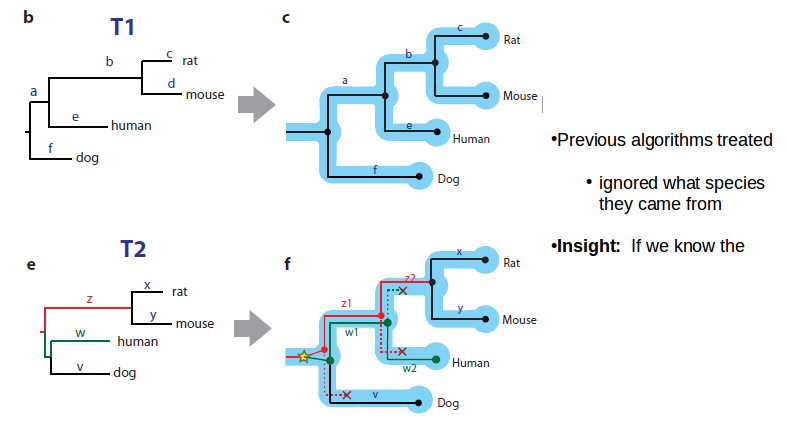
If we know the species tree, we can develop a model for what kind of branch lengths we can expect. We can use conserved gene order to tell orthologs and build trees.

When a gene is fast evolving in one species, it is fast evolving in all species. We can model a branch length as two different rate components. One is gene specific (present across all species) and the other is species specific, which is customized to a specific species.
%253D613.190.png?revision=1)
This method greatly improves reconstruction accuracy.