22.3: Molecular Methods for Studying Nuclear Genome Organization
- Page ID
- 41052

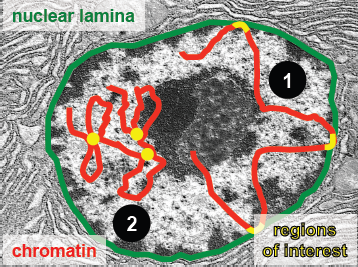
There are two main types of methods for investigating the three-dimensional structure of chromatin in the nucleus.
- The first set of methods, ChIP and DamID, are methods that measure DNA-’landmark’ interactions. That is, they measure interactions of genome loci with relatively fixed nuclear landmarks, and only regions of the genome that come into contact with the nuclear lamina will be identified.
The second set of methods, the 3C-based methods, are those that measure DNA-DNA interactions. Any two regions of DNA that interact may be identified, regardless of whether they are near the interior or periphery of the nucleus.
Methods for measuring DNA-Nuclear Lamina interactions
The following methods, ChIP and DamID, both examine regions of DNA the specifically come in contact with the nuclear lamina.
ChIP: Chromatin Immuno Precipitation
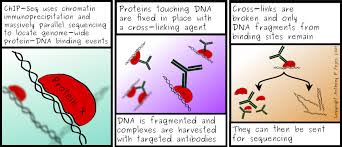
ChIP is a method for detecting regions of DNA that are bound to proteins of interest. Proteins bound with DNA are cross-linked in place with formaldehyde. The protein-DNA complexes are pulled-down using anity chromatography, mainly using specific antibodies that target the protein of interest. The recovered complexes are then dessassociated, cross-links are broken, and the DNA that was bound to the proteins is fragmented and analyzed. DNA fragments can be then analyzed using sequencing (ChiP-Seq) or microarrays (ChiP-Chip). However, a big challenge associated with the various ChIP techniques is that it can be dicult to get a high-anity antibody. To study the 3D structure of DNA within the nucleus, ChIP-Seq can be used with antibodies that targed lamina proteins.
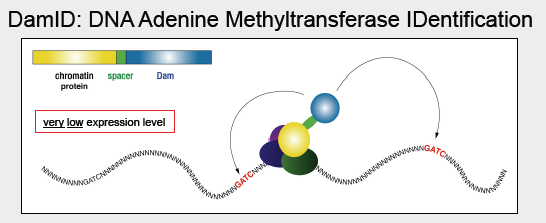
DamID: DNA adenine methyltransferase IDentification
DamID is used to map the binding sites of Chromatin-binding proteins. In the DamID method, DNA adenine methyltransferase (Dam) from E. coli is fused to the LaminB1 protein (the Dam enzyme hangs off the end of the protein and is thus in the vicinity for interactions). In E. coli, the Dam enzyme methylates the adenine in the sequence GATC; bacterial genomes contain proteins with functions like Dam to protect their own DNA from digestion by restriction enzymes, or as part of their DNA repair systems. As this process doesnt naturally occur in eukaryotes, the methylated adenines in a region can thus be attributed to an interaction with the protein fused with Dam, thereby implying that that particular region came into close contact with the nuclear lamina. As a control, unfused Dam can be expressed at low levels. This results in a sparse distribution of methylated adenine for which the precise position of the methylated adenines can be used to infer variation in DNA accessibility. The methylated adenine are determined using disulphide PCR assays or other PCR technique sensitive to methylations in the template DNA. In one of those assays, the genome can digested by DpnI, which only cuts methylated GATC sequences. Adapter sequences are then ligated to the ends of these digested pieces, and PCR is run using primers matching the adapters. Only the regions occurring between proximal GATC positions are amplified. The final measurement is the log of the ratio between test and control lamina association: positive values are preferentially lamina-associated, and are thus identified as LADs. One advantage of using DamID over ChIP is that DamID does not require a specific antibody which may be dicult to find. However, a disadvantage of using DamID is that the fusion protein must be made and expressed.
Courtesy of Anthony P. Fejes. Used with permission.
Courtesy of Bas van Steense. Used with permission.
FAQ
Q: How close does DNA have to come to DamID to be methylated?
A: It doesn’t have to bind directly to the lamina, but it does have to come pretty close. DamID has a range of about 1.5kb.
Measuring DNA-DNA contacts
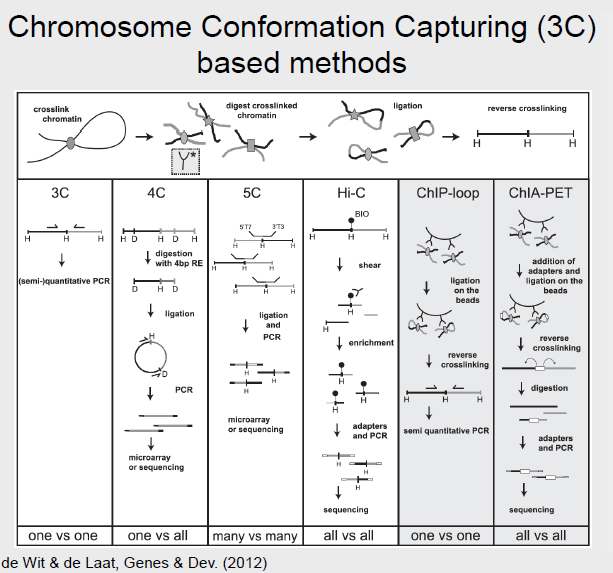
All of the following methods are based on Chromosome Conformation Capturing (3C) with certain modifications.
Cold Spring Harbor Laboratory Press. All rights reserved. This content is excluded from our Creative
Commons license. For more information, see http://ocw.mit.edu/help/faq-fair-use/.
Source: de Wit, Elzo and Wouter de Laat. "A decade of 3C Technologies: Insights into Nuclear Organization."
Genes & Development 26, no. 1 (2012): 11-24.
3C
Chromosome Conformation Capturing (3C) is a method that detects which genomic loci are in close vicinity to other loci within the nucleus. Similar to the ChIP method, a cross-linking agent is used freeze proteins bound to DNA in place and forming protein-DNA complexes. The DNA can be then be digested by a restriction enzyme after allowing the bound protein to disassociate. Typically an enzyme with a 6 bps long recognition site that leaves sticky ends, like HindIII, is used. The generated fragments are then induce to self-ligate (Using very low concentration of DNA to prevent the ligation of the fragment with another random fragment). The result is a pool of linear DNA fragments, known as the 3C library, that may be analyzed via PCR by designing primers specifically for the interaction of interest. 3C can be described as a ’one vs one’ method, because the primers used are specifically target to amplify the product of the interaction between 2 regions of interest.
Circularized Chromatin Conformation Capture (4C)
4C methods can be described as a ’one vs all’ because for a single region of interest, we can examine all its interactions with all other regions in the genome. 4C works similarly to 3C with the main di↵erence being the restriction enzyme used. In 4C, a common cutter is employed to generate more and smaller fragments. These fragments are then again ligated. Some smaller fragments may be excluded, but the result is a circularized fragment of DNA. Primers can be designed to amplify the ’unknown’ fragment of DNA so that all interactions with the region of interest are identified.
Carbon-Copy Chromosome Conformation Capture (5C)
5C is a ’many vs many’ method and allows the identification of interactions between many regions of interest and many other regions, also of interest, to be analyzed at once. 5C works similarly to 3C. However, after obtaining the 3C library, multiplex ligation-mediated amplification (LMA) is performed. LMA is a method in which multiple targets are amplified. The resulting 5C library may be analyzed on a microarray or high-throughput sequencing.
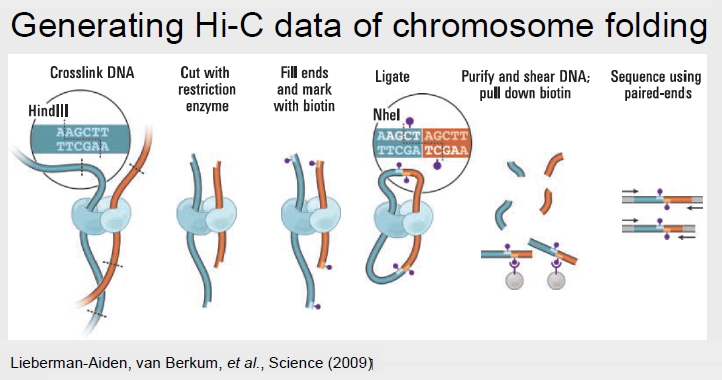
Hi-C
Hi-C can be described as an ’all vs all’ method because it identifies all chromatin interactions. Hi-C works by labeling all DNA fragments with biotin before ligation, which marks all the ligation junctions. Magnetic beads are then used to purify the biotin-marked junctions. This Hi-C library may then be fed into next generation sequencing.
ChIP-loop
ChIP-loop can be described as a ’one vs one’ method, because similar to 3C, only an interaction between two regions of interest may be identified. ChIP-loop is a hybrid between ChIP and the 3C methods. DNA-protein complexes are first cross-linked and digested. Then, as in ChIP, the protein of interest and the DNA bound to it are pulled down using an antibody. The protocol then proceeds as in 3C: the free ends of the fragments are ligated, the cross-linking are reversed, and sequencing can proceed using primers designed specifically for a ’one vs one’ interaction.
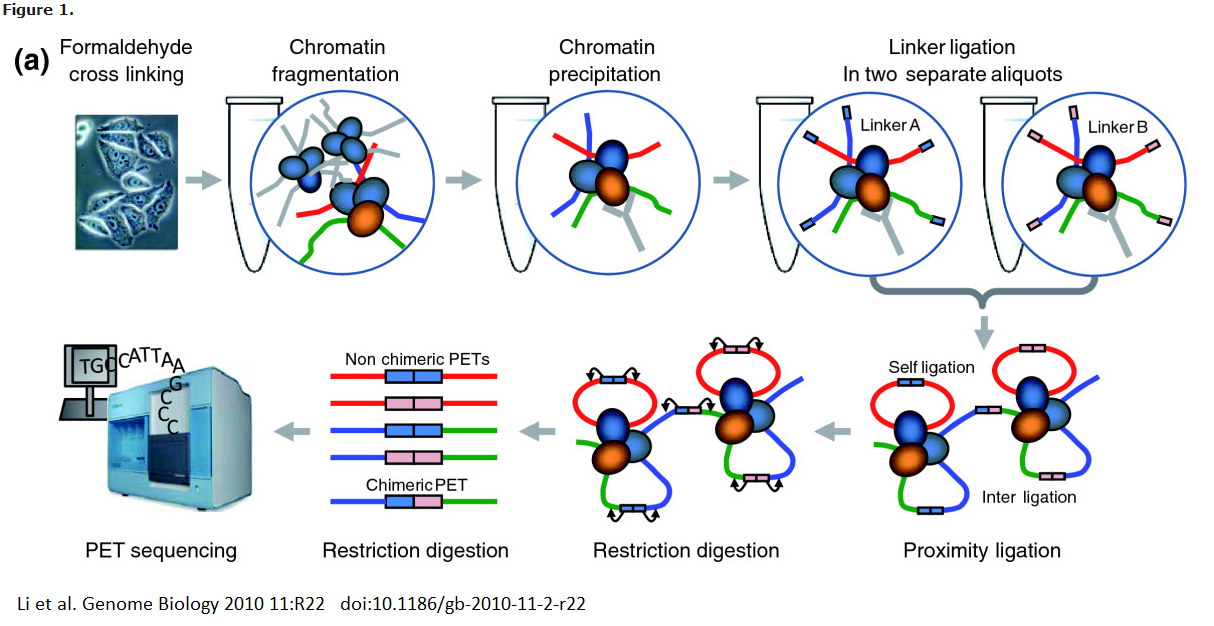
ChIA-PET
Chromatin Interception Analysis by Paired-End Tag Sequencing, or ChIA-PET, combines the ChIP and 3C methodologies to determine long-range chromatin interactions genome-wide. It can be described as a ’all vs all’ method, because although a single protein of interest must be identified, any interactions will be identified. In ChIA-PET, DNA-protein complexes are cross-linked, as in previously discussed methods. However, sonication is then used to break up chromatin, and to reduce non-specific interactions. As in the ChIP protocol, an antibody is used to pull down regions of DNA bound to a protein of interest. Two di↵erent oligonucleotide linkers are then ligated to the free ends of the DNA. These linkers both have MmeI cut sites. The linkers are then ligated together so that the free ends are connected, after which the fragments are digested with MmeI. MmeI cuts 20 nt downstream of its recognition sequence, so the result of the digestion is the linker bordered by the sequence of interest on either side. This is a ’tag-linker-tag’ structure, and the fragments are known as PETs. The PETs may be sequenced and mapped back to the genome to determine regions of interacting DNA.