
11.2: Post-Transcriptional Regulation
- Page ID
- 40983

Basics of Protein Translation
For the basics of transcription and translation, refer to Lecture 1, sections 4.3 - 4.5.

The genetic code is almost universal.
|
FAQ Q: Why is genetic code so similar across organisms? A: Genomic material is not only transmitted vertically (from parents) but also horizontally between organisms. This gene interaction creates an evolutionary pressure for an universal genetic code. |
|
FAQ Q: What accounts for the slight differences in the genetic code across organisms? A: Late/early evolutionary arrival of amino acids can account for the differences. Also, certain species (e.g. bacteria in deep sea vents) have more resources to synthesize specific amino acids, thus they will favor those in the genetic code. |
|
Did You Know? Threonine and Alanine are often accidentally interchanged by tRNA sythetase because they originated from one amino acid. |
Measuring Translation
Translation efficiency is defined as,
\[T_{e f f}=\frac{[\mathrm{mRNA}]}{[\mathrm{protein}]}\nonumber\]
We are interested in seeing just how much of our mRNA is translated to protein, i.e. the efficiency. However, specifically measuring how much mRNA becomes protein is a difficult task, one that requires a bit of creativity. There are a variety of ways to tackle this problem, but each has its own downfalls:
1. Measure mRNA and protein levels directly
Pitfall: Does not consider rates of synthesis and degradation. This method measures the protein levels for the ’old’ mRNA since there is a time lag from mRNA to protein.
2. Use drugs to inhibit transcription and translation Pitfall: Drugs have side effects altering translation
3. Artificial fusion of proteins with tags
Pitfall: Protein tags can affect protein stability
4. Pulse label with radioactive nucleosides or amino acids (SILAC) **in use today**
Pitfall: Offers no information on dynamic changes: it is simply a snapshat of the resulting mRNA and protein levels after X hours 193
Another common technique is using ’ribosome profiling’ to measure protein translation at subcodon res- olution. This is done by freezing ribosomes in the process of translation and degrading the non-ribosome protected sequences. At this point, the sequences can be pieced back together and the frequency with which a region is translated can be interpolated. The disadvantage to using these ribosome footprints, to see which regions are being translated, is that regions in between ribosomes are lost. This technique requires an RNA-seq in parallel.
The question remains, why is Ribosome profiling advantageous? This technique is a better approach to measuring protein abundance as it:
1. Is a better measure of protein abundance
2. Is independent of protein degradation (compared to the protein abundance/mRNA ratio)
3. Allows us to measure codon-specific translation rates
Using ribosome profiling, it is possible to see which codon is being decoded: this is done by mapping ribosome footprints and then deciphering the translating codon based on footprint length. We can the verify our prediction by mapping translated codon profiles based on periodicity (three bases in a codon). The technique can be improved even further by using anti-translation drugs such as harringtonine and cyclohexamide. Cyclohexamide blocks elongation and Harringtonine inhibits initiation. The later can be used to find the starting points (which genes are about to be translated). Figure 4 shows the effects of the drugs on the ribosome profiles.
This technique has much more to offer than simply quantifying translation. Ribosome profiling allows for:
1. Prediction of alternative isoforms (different places where translation can start) images/AltIsoforms.png

2. Prediction of un-indentified ORFs (open reading frames)
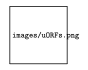
Figure 11.6: Ribosome profile when harringtonine is used vs. no drug. The red peaks previously un-identified ORFs.
3. Comparing translation across different environmental conditions

4. Comparing translation across life stages
Thus, we see that ribosome profiling is a very powerful tool with lots of potential to reveal previously elusive information about the translation of a genome.
Codon Evolution
Basic concepts
Something to make clear is that codons are not used with equal frequencies. In fact, which codons can be considered optimal differs across different species based on RNA stability, strand-specific mutation bias, transcriptional efficacy, GC composition, protein hydropathy, and translational efficiency. Likewise, tRNA isoacceptors are not used with equal frequencies within and across species. The motivation for the next section is to determine how we may measure this codon bias.
Measures of Codon Bias
There are a few methods to accomplish this task:
a) Calculate the frequency of optimal codons, which is defined as “optimal” codons/ sum of “optimal” and “non-optimal” codons. The limitations to this method are that this requires knowing which codon is recognized by each tRNA and it assumes that tRNA abundance is highly correlated with tRNA gene copy number.
b) Calculate a codon bias index. This measures the rate of optimal codons with respect to the total codons encoding for that same amino acid. However, in this case the number of optimal codons are normalized with respect to the expected random usage. CBI = (oopt − erand)/(otot − erand). The limitation of this method is that it requires a reference set of proteins, such as highly expressed ribosomal proteins.
c) Calculate a codon adaptation index. This measures the relative adaptiveness or deviation of the codon us- age of a gene towards the codon usage of a reference set of proteins, i.e. highly expressed genes. It is defined as the geometric mean of the relative adaptiveness values, measured as weights associated to each codon over the length of the gene sequence (measured in codons). Each weight is computed as the ratio between the observed frequency of a given codon and the frequency of its corresponding amino acid. The limita- tion to this approach is that it requires the definition of a reference set of proteins, just as the last method did.
d) Calculate the effective number of codons. This measures the total number of different codons used in a sequence, which measures the bias toward the use of a smaller subset of codons, away from equal use of synonymous codons. Nc = 20 if only one codon is used per amino acid, and Nc = 61 when all possible synonymous codons are used equally. The steps to the process are to compute the homozygosity for each amino acid as estimated from the squared codon frequencies, obtain effective number of codons per amino acid, and compute the overall number of effective codons. This method is advantageous because it does not require any knowledge of tRNA-codon pairing, and it does not require any reference set However, it is limited in that it does not take into account the tRNA pool.
e) Calculate the tRNA adaptation index. Assume that tRNA gene copy number has a high positive correla- tion with tRNA abundance within the cell. This then measures how well a gene is adapted to the tRNA pool.
It is important to distinguish among when to use each index. The situation in which a certain index is favorable is very context-based, and thus it is often preferable to use one index above all others when the situation calls for it. By carefully choosing an index, one can uncover information about the frequency by which a codon is translated to an amino acid.
RNA Modifications
The story becomes more complicated when we consider modifications that can occur to RNA. For instance, some modifications can expand or restrict the wobbling capacity of the tRNA. Examples include insosine modifications and xo5U modifications. These modifications allow tRNAs to decode a codon that they could not read before. One might ask why RNA modification was positively selected in the context of evolution, and the rationale is that this allows for the increase in the probability that a matching tRNA exists to decode a codon in a given environment.
Examples of applications
There are a few natural applications that result form our understanding of codon evolution.
a) Codon optimization for heterologous protein expression
b) Predicting coding and non-coding regions of a genome
c) Predicting codon read-through
d) Understanding how genes are decoded - studying patterns of codon usage bias along genes
Translational Regulation
There are many known means of regulation at the post-transcriptional level. These include modulation of tRNA availability, changes in mRNA, and cis -and trans-regulatory elements. First, tRNA modulation has a large impact. Changes in tRNA isoacceptors, changes in tRNA modifications, and regulation at tRNA aminoacylation levels. Changes in mRNA that affect translation include changes in mRNA modification, polyA tail, splicing, capping, and the localization of mRNA (importing to and exporting from nucleus). Cis- and trans- regulatory elements include RNA interference (i.e. siRNA and miRNA), frameshift events, and riboswitches. Additionally, many regulatory elements are still yet to be discovered!