
7.5: DNA Lesions
- Page ID
- 16132

The robust nature of DNA due to its complementary double strands has been noted several times already. We now consider in more detail the repair processes that rescue damaged DNA. DNA is not nearly as robust as popular media makes it out to be. In fact, to take the blockbuster book and film, Jurassic Park, as an example, Although there is unquestionably some DNA to be found either embedded in amber-bound parasites, or perhaps in preserved soft tissue (found deep in a fossilized femur, Schweitzer et al, 2007). It is likely to be heavily degraded, and accurate reproduction is impossible without many samples to work from.
The most common insult to the DNA of living organisms is depurination, in which the β-N-glycosidic bond between an adenine or guanine and the deoxyribose is hydrolyzed. In mammalian cells, it is estimated at nearly 10000 purines per cell generation, and generally, the average rate of loss at physiological pH and ionic strength, and at 37°C, is approximately 3 x 10-11 /sec. Depyrimidination of cytosine and thymine residues can also occur, but do so at a much slower rate than depurination. Despite the high rate of loss of these bases, they are generally remediated easily by base excision repair (BER), which is discussed later in this section. Therefore it is rare for depurination or depyrimidination to lead to mutation.
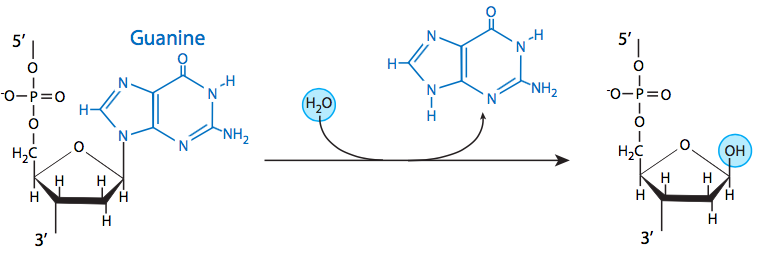
Three of the four DNA bases, adenine, guanine, and cytosine, contain amine groups that can be lost in a variety of pH and temperature-dependent reactions that convert the bases to hypoxanthine, xanthine, and uracil, respectively. This can sometimes lead to permanent mutations since during replication, they serve as a template for the synthesis of a complementary strand, and where a guanine should go, for example (complementary to cytosine), an adenine may be inserted (because it complements uracil, the deamination product of cytosine).
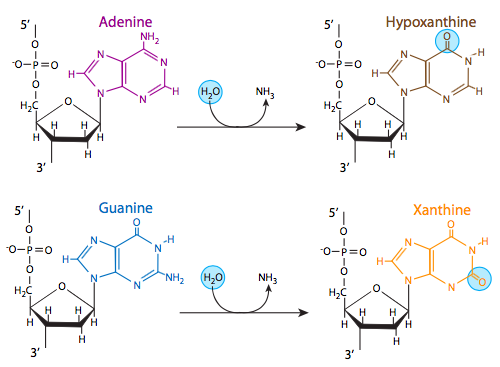
Another deamination, of the modified base methylcytosine, can also lead to a mutation upon replication. Some cytosines may be methylated as part of a regulatory process to inactivate certain genes in eukaryotes, or in prokaryotes as protection against restriction endonucleases. When the methylated cytosine is deaminated, it produces a thymine, which changes the complementary nucleotide (upon replication) from a guanine to an adenine. Deamination of cytosines occurs at nearly the same rate as depurination, but deamination of other bases are not as pervasive: deamination of adenines, for example, is 50 times less likely than deamination of cytosine.

Thymine good, Uracil bad. Why is thymine found in DNA rather than uracil? It turns out that the frequency of cytosine deamination may yield a clue as to why cells have gone the extra step (literally, since uracil is a precursor in thymine biosynthesis) to make a new “standard” nucleotide for DNA when uracil worked just fine for RNA, presumably the older genetic molecule. Consider this: if uracil was standard for DNA, then the very frequent deamination conversions of C to U would not be caught by error-checking for non-DNA bases, and the mutation rate would skyrocket. Fortunately, since T has evolved to be the standard base-pairing partner of adenine in DNA, uracil is quickly recognized and removed by multiple uracil DNA glycosylases (more on that later in this chapter), and the integrity of our DNA sequences is much safer.
All DNA bases can spontaneously shift to a tautomeric isomer (amino to imino, keto to enol, etc), although equilibrium leans heavily toward one than the other. When a rare tautomer occurs, it base-pairs differently than its more common structural form: guanines with thymines and adenines with cytosines. Here again, a mutation can be propagated during replication of the DNA.
DNA inside a cell must also contend with reactive oxidative species (ROS) generated by the cell’s metabolic processes. These include singlet oxygen, peroxide and peroxide radicals, as well as hydroxyl radicals. although it is thought that the hydrogen peroxide and peroxide radicals do not directly attack the DNA but rather generate hydroxyl radicals that do. Most of these ROS are generated in the mitochondria during oxidative phosphorylation and leak out, although some may be generated in peroxisomes, or in some cytosolic reactions. Depending on what part of the DNA is targeted, ROS can cause a range of lesions including strand breaks and removal of bases.
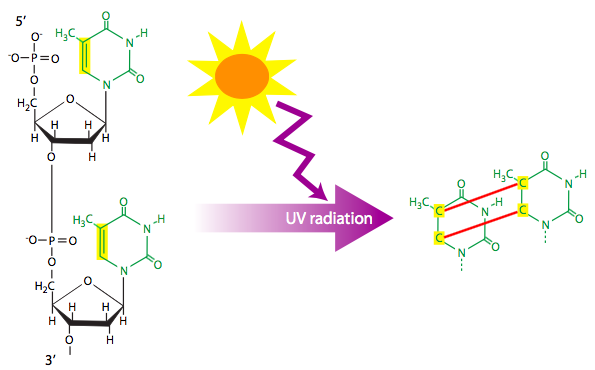
Ionizing radiation (e.g. X-rays) and ultraviolet radiation can each cause DNA lesions. Ionizing radiation is often a cause for double-stranded breaks of the DNA. As described later in the chapter, the repair process for double-stranded breaks necessarily leads to some loss of information, and could potentially knock out a gene. Ultraviolet radiation that hits adjacent thymines can cause them to react and form a cyclobutyl (four carbons bonded in closed loop) thymine dimer. The dimer pulls each thymine towards the other, out of the normal alignment. Depending on the structural form of the dimer, this is sufficient to stymie the replication machine and halt replication. However, some data suggests that normal base-pairing to adenine may be possible under some conditions, although, it is likely only one base-pair would result, and the missing base could lead to either random substitution or a deletion in the newly synthesized strand.
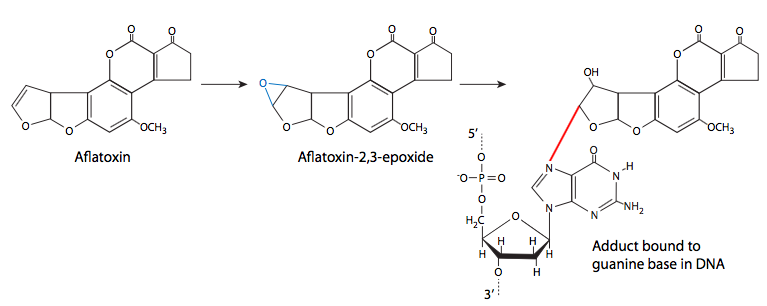
Finally, we consider the formation of chemical adducts (covalently attached groups) on DNA. They may come from a variety of sources, including lipid oxidation, cigarette smoke, and fungal toxins. These adducts attach to the DNA in different ways, so there are a variety of different effects from the adducts as well. Some may be very small adducts - many environmental carcinogens are alkylating agents, transferring methyl groups or other small alkyl groups to the DNA. Other adducts are larger, but also attach covalently to a nitrogenous base of DNA. Common examples are benzo(a)pyrene, a major mutagenic component of cigarette smoke, and aflatoxin B1, produced by a variety of Aspergillus-family fungi. Benzo(a)pyrene is converted to benzo(a)pyrene diol epoxide, which can then attack the DNA. When this happens, the at pyrene ring intercalates between bases, causing steric changes that lead to local deformation of the DNA and disruption of normal DNA replication.

Aflatoxin B1 is the primary aflatoxin produced by some species (esp. flavus, parasiticus) of Aspergillus, a very common mold that grows on stored grain (as well as detritus and other dead or dying plant matter). In addition to infecting grain, it is a common problem with stored peanuts. At high levels, aflatoxin is acutely toxic, but at lower levels, it has the insidious property of being unnoticeably toxic but mutagenic. Like benzo(a) pyrene, it is metabolized into an epoxide and will then react with DNA to form an adduct that can disrupt replication.
Some alkylating agents, particularly N-nitroso compounds, are formed in the acidic conditions of the stomach from nitrosation of naturally occurring nitrites produced from food (reduction of nitrates), or environmental nitrites in drinking water. Ironically, while some alkylating agents can cause cancers, others are used therapeutically as anticancer treatments, e.g. mitomycin, melphalan. The idea, as with many cancer treatments, is that although such drugs cause DNA damage to non-cancerous cells as well as cancer cells, the high rate of cancer cell proliferation gives them fewer chances for repair of damaged DNA, and thus greater likelihood that the damage might halt replication and lead to cell death.
In a similar vein, crosslinking chemotherapeutic agents such as cisplatin (a platinum atom bonded to two chloride groups and two amino groups) also bind to DNA. The chloride groups are displaced first by water and then by other groups including sites on DNA. Although sometimes classified as an alkylating agent, it obviously is not, but it acts similarly. Cisplatin goes a step further than a simple alkylating agent though, because it has another reactive site and can thus crosslink (covalently bond) another nucleotide, possibly on another strand of DNA, making a strong obstruction to DNA replication. Cisplatin can also crosslink proteins to DNA.
Benzo(a)pyrene and aflatoxin B1 are not themselves mutagens. Once they are in the cell, the normal metabolism of these compounds leads to diol epoxide formation, which can then attack the DNA. Although the 7-nitrogen (N7) of guanine is more nucleophilic, and is a target for aflatoxin, most benzo(a)pyrene diol epoxide adducts attach to the 2-nitrogen of guanine residues.
There are federal standards (20-300 parts per billion depending on usage) for aflatoxin in various forms of grain-based animal feed, especially corn-based feeds, because the toxin can pass through the animal into milk, as well as linger in the meat. In addition to feed, there are federal maximums for peanuts and peanut products, brazil nuts, pistachios, and other foodstuffs (actionable at 20 ppb).
Well then, what’s a poor cell to do when its DNA is being constantly ravaged? As it turns out, there are some very good repair processes that are constantly at work on the DNA, scanning it for defects, and where possible, making repairs. Often the repairs are perfect, if the complementary strand is intact, sometimes mutations must be introduced, and finally there are occasions when repair is impossible, and apoptosis is triggered to kill the cell and prevent propagation of damaged DNA.