
7.3: Prokaryotic Replication
- Page ID
- 16130

DNA replication begins at an origin of replication. There is only one origin in prokaryotes (in E. coli, oriC) and it is characterized by arrays of repeated sequences. These sequences wrap around a DNA-binding protein, and in doing so, exert pressure on the H-bonds between the strands of DNA, and the chromosome begins to unzip in an AT-rich area wrapped around this protein. Remember that A-T pairs are 33% weaker than G-C pairs due to fewer hydrogen bonds. The use of AT-rich stretches of DNA as points of strand separation is a recurring theme through a variety of DNA operations. The separation of the two strands is bidirectional, and DNA polymerases will act in both directions in order to finish the process as quickly as possible. Speed is important here because while replication is happening, the DNA is vulnerable to breakage, and most metabolic processes are shut down to devote the energy to the replication. Even in prokaryotes, where DNA molecules are orders of magnitude smaller than in eukaryotes, the size of the DNA molecule when it is unraveled from protective packaging proteins makes it highly susceptible to physical damage just from movements of the cell.
The first OriC binding protein, DnaA, binds to DnaA boxes, which are 9 base pair segments with a consensus sequence of TTATCCACA. OriC has five of these repeats, and one DnaA protein binds to each of them. HU and IHF are histone-like proteins that associate with DnaA and together bend that part of the DNA into a circular loop, situating it just over the other major feature of oriC, the 13-bp AT-rich repeats (GATCTNTTNTTTT). DnaA hydrolyzes ATP and breaks the H-bonds between strands in the 13mer repeats, also known as melting the DNA. This allows complexes of DnaB [and DnaC, which is a loading protein that helps attach DnaB(6) to the strand with accompanying hydrolysis of ATP. Also, five more DnaA are recruited to stabilize the loop.] to bind to each single-stranded region of the DNA on opposite sides of the newly opened replication bubble.
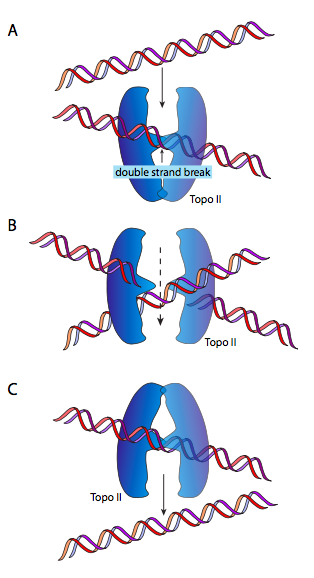
DnaB is a helicase; its enzymatic activity is to unzip/unwind the DNA ahead of the DNA polymerase, to give it single-stranded DNA to read and copy. It does so in association with single-stranded-DNA binding proteins (SSBs), and DNA gyrase. The function of SSB is nearly self-explanatory: single-stranded DNA is like RNA in its ability to form complex secondary structures by internal base-pairing, so SSB prevents that. DNA gyrase is a type II topoisomerase, and is tasked with introducing negative supercoiling to the DNA. This is necessary because the unzipping of the DNA by helicase also unwinds it (since it is a double helix) and causes the introduction of positive supercoiling. This means that the entire circular molecule twists on itself: imagine holding a rubber band in two hands and twisting it. As the supercoiling accumulates, the DNA becomes more tightly coiled, to the point that it would be impossible for helicase to unzip it. DnaB/gyrase can relieve this stress by temporarily cutting the double-stranded DNA, passing a loop of the molecule through the gap, and resealing it.
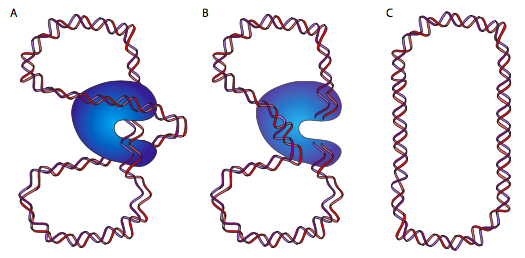
This (hopefully) makes a lot more sense looking at the diagram. Or, going back to our rubber band, give the rubber band a twist or two, then tape down the two ends. If you snip the rubber band, and pass an adjacent portion of the rubber band through that snip, then reconnect the cut ends, you will find that there is one less twist. Nifty, eh? At this point, some of you are going to say, but if you twist a free-floating rubber band, as one might imagine a free-floating circular DNA chromosome in E. coli, you would expect it to naturally untwist. Technically, yes, but due to the large mass of the chromosome, its association with various proteins and the cell membrane, and the viscosity of its environment, it does not behave as though it were completely free.
Once oriC has been opened and the helicases have attached to the two sides of the replication fork, the replication machine, aka the replisome can begin to form. However, before the DNA polymerases take positions, they need to be primed. DNA polymerases are unable to join two individual free nucleotides together to begin forming a nucleic acid; they can only add onto a pre-existing strand of at least two nucleotides. Therefore, a specialized RNA polymerase (RNAP’s do not have this limitation) known as primase is a part of the replisome, and reads creates a short RNA strand termed the primer for the DNA polymerase to add onto. Although only a few nucleotides are needed, the prokaryotic primers may be as long as 60 nt depending on the species.
At least five prokaryotic DNA polymerases have been discovered to date. The primary DNA polymerase for replication in E. coli is DNA Polymerase III (Pol III). Pol I is also involved in the basic mechanism of DNA replication, primarily to ll in gaps created during lagging strand synthesis (defined 3 pages ahead) or through error-correcting mechanisms. DNA polymerase II and the recently discovered Pol IV and Pol V do not participate in chromosomal replication, but rather are used to synthesize DNA when certain types of repair is needed at other times in the cellular life cycle.
DNA polymerase III is a multi-subunit holoenzyme, with α, ε, and θ subunits comprising the core polymerase, and τ, γ, δ, δ’, χ, Ψ, and β coming together to form the complete holoenzyme. The core polymerase has two activities: the α subunit is the polymerase function, reading a strand of DNA and synthesizing a complementary strand with great speed, around 150 nt/sec; the ε subunit is a 3’-5’ “proofreading” exonuclease and acts as an immediate proofreader, removing the last nucleotide if it is incorrect. This proof- reading does not reach any further back: it only acts on the most recently added nucleotide to correct misincorporation. Other mechanisms and enzymes are used to correct DNA lesions that arise at other times. [As a matter of nomenclature, exonucleases only cut off nucleotides from DNA or RNA from either end, but not in the middle. Endonucleases cleave phosphodiester bonds located deeper within a nucleic acid strand.] The θ subunit has no enzymatic activity and regulates the exonuclease function. Although it has polymerase activity, the Pol III core polymerase has poor processivity - that is, it can only add up to 15 nucleotides before dissociating from the template DNA. Since genomes of E. coli strains average near 5 million base pairs, replication in little 15 nt segments would be extraordinarily inefficient.
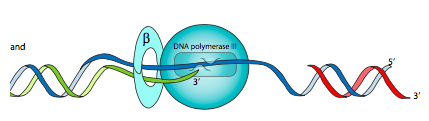
The clamp loader complex is an ATPase assembly that binds to the β-clamp unit upon binding of ATP (but the ATPase activity is not turned on). When the complex then binds to DNA, it activates the ATPase, and the resulting hydrolysis of ATP leads to conformational changes that open up the clamp temporarily (to encircle or to move off of the DNA strand), and then dissociation of the clamp loader from the clamp assembly.
This is where the β subunit is needed. Also known as the β clamp, it is a dimer of semicircular subunits that has a central hole through which the DNA is threaded. The core polymerase, via an α-β interaction, is attached to this β clamp so that it stays on the DNA longer, increasing the processivity of Pol III to over 5000nt. The β clamp is loaded onto (and unloaded off of) the DNA by a clamp loader complex (also called γ complex) consisting of γ (x3), δ, δ’, χ, and Ψ subunits.
The replication bubble has two replication forks - once the DNA is opened up (unzipped) at the origin, a replication machine can form on each end, with the helicases heading in opposite directions. For simplification, we will consider just one fork — opening left to right — in this discussion with the understanding that the same thing is happening with the other fork, but in the opposite direction.
The first thing to notice when looking at a diagram of a replication fork (Figure \(\PageIndex{11}\)) is that the two single-stranded portions of template DNA are anti-parallel. This should come as no surprise at this point in the course, but it does introduce an interesting mechanical problem. Helicase opens up the double stranded DNA and leads the rest of the replication machine along. So, in the single-stranded region trailing the helicase, if we look left to right, one template strand is 3’ to 5’ (in blue), while the other is 5’ to 3’ (in red). Since we know that nucleic acids are polymerized by adding the 5’ phosphate of a new nucleotide to the 3’ hydroxyl of the previous nucleotide (5’ to 3’, in green), this means that one of the strands, called the leading strand, is being synthesized in the same direction that the replication machine moves. No problem there.
The other strand is problematic: looked at linearly, the newly synthesized strand would be going 3’ to 5’ from left to right, but DNA polymerases cannot add nucleotides that way. How do cells resolve this problem? A number of possibilities have been proposed, but the current model is depicted here. The replication machine consists of the helicase, primases, and two DNA polymerase III holoenzymes moving in the same physical direction (following the helicase). In fact, the pol III complexes are physically linked through τ subunits.
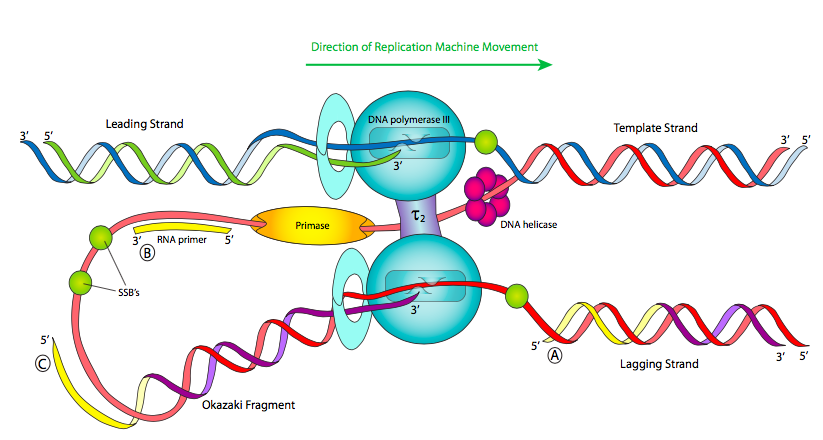
In order for the template strand that is 5’ to 3’ from left to right to be replicated, the strand must be fed into the polymerase backwards. This can be accomplished either by turning the polymerase around or by looping the DNA around. As the Figure shows, the current model is that the primase is also moving along left to right, so it has just a short time to quickly synthesize a short primer before having to move forward with the replisome and starting up again, leaving intermittent primers in its wake. Because of this, Pol III is forced to synthesize only short fragments of the chromosome at a time, called Okazaki fragments after their discoverer. Pol III begins synthesizing by adding nucleotides onto the 3’ end of a primer and continues until it hits the 5’ end of the next primer. It does not (and can not) connect the strand it is synthesizing with the 5’ primer end.
DNA replication is called a semi-discontinuous process because while the leading strand is being synthesized continuously, the lagging strand is synthesized in fragments. This leads to two major problems: first, there are little bits of RNA left behind in the newly made strands (just at the 5’ end for the leading strand, in many places for the lagging); and second, Pol III can only add free nucleotides to a fragment of single stranded DNA; it cannot connect another fragment. Therefore, the new “strand” is not whole, but riddled with missing phosphodiester bonds.
The first problem is resolved by DNA polymerase I. Unlike Pol III, Pol I is a monomeric protein and acts alone, without additional proteins. There are also 10-20 times as many Pol I molecules as there are Pol III molecules, since they are needed for so many Okazaki fragments. DNA Polymerase I has three activities: (1) like Pol III, it can synthesize a DNA strand based on a DNA template, (2) also like Pol III, it is a 3’-5’ proofreading exonuclease, but unlike Pol III, (3) it is also a 5’-3’ exonuclease. The 5’-3’ exonuclease activity is crucial in removing the RNA primer (Figure \(\PageIndex{12}\)). The 5’-3’ exonuclease binds to double- stranded DNA that has a single-stranded break in the phosphodiester backbone such as what happens after Okazaki fragments have been synthesized from one primer to the next, but cannot be connected. This 5’-3’ exonuclease then removes the RNA primer. The polymerase activity then adds new DNA nucleotides to the upstream Okazaki fragment, filling in the gap created by the removal of the RNA primer. The proofreading exonuclease acts just like it does for Pol III, immediately removing a newly incorporated incorrect nucleotide. After proofreading, the overall error rate of nucleotide incorporation is approximately 1 in 107.
Technically, the 5’-3’ exonuclease cleaves the DNA at a double-stranded region downstream of the nick, and may then remove anywhere from 1-10nt at a time. Experimentally, the 5’-3’ exonuclease activity can be cleaved from the rest of Pol I by the protease trypsin. This generates the “Klenow fragment” containing the polymerase and 3’-5’ proofreading exonuclease.
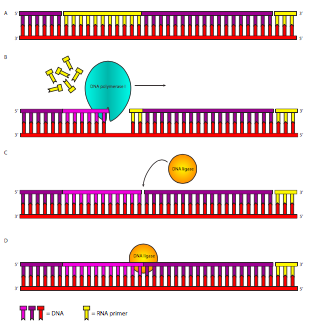
Figure \(\PageIndex{12}\). Lagging Strand Synthesis. After DNA polymerase III has extended the primers (yellow), DNA polymerase I removes the primer and replaces it by adding onto the previous fragment. When it finishes removing RNA, and replacing it with DNA, it leaves the DNA with a missing phosphodiester bond between the pol III-synthesized DNA downstream and the pol I-synthesized DNA upstream. This break in the sugar-phosphate backbone is repaired by DNA ligase.
Even though the RNA has been replaced with DNA, this still leaves a fragmented strand. The last major player in the DNA replication story finally appears: DNA ligase. This enzyme has one simple but crucial task: it catalyzes the attack of the 3’-OH from one fragment on the 5’ phosphate of the next fragment, generating a phosphodiester bond. This reaction requires energy in the form of hydrolysis of either ATP or NAD+ depending on the species (E. coli uses NAD+) generating AMP and either PPi or NMN+.