16.9: DNA Rearrangement
- Page ID
- 19400
One of the central assumptions throughout our study of the cell has been that although the RNA and proteins in any cell may differ, any cell of a given organism other than the gametes should have the same DNA. This is not the case with B cells or T cells. In these cells, part of the maturation process is to create a unique arrangement of different domains to form a specific antibody (or T-cell receptor). The germline DNA, or the DNA that is found in all other somatic cells of the organism, contains many different such segments, but only a few are put together to make the antibody/TCR. This is a stochastic process, and with this kind of rearrangement happening on both heavy chain genes and two different light chain genes (designated κ and λ), there are well over 10 trillion (1013) different combinations for generating immunoglobulins in humans, and even more combinations for T-cell receptors! How is this accomplished?
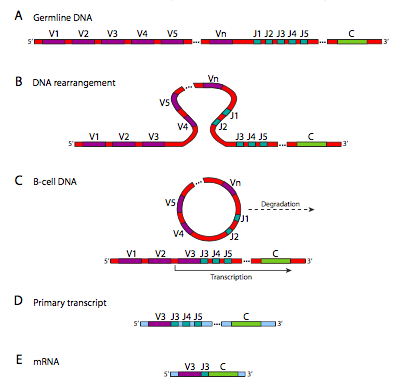
Figure \(\PageIndex{19}\) shows the DNA rearrangements that take place in generating κ chain diversity. The λ chain locus has a slightly different arrangement, and has only 30 V genes, with 4 J segments and 4 C genes. The heavy chain has an extra domain: there are 40 V genes, which are linked to one of 25 D segments, then 6 potential J segments, and one C gene. These rearrangements, although they look something like the RNA splicing that we saw earlier in this course, are happening at the DNA level. Once it has happened, that cell and its progeny can no longer make the other combinations because those parts of the genome have been cut out and destroyed. This is distinctly different from alternative splicing of RNA, in which the genetic information is still there, and under different conditions could still generate other variations of the gene product.

The enzyme that produces this diversity is a complex called the V(D)J recombinase. The recombination occurs in two parts: first double-stranded breaks are made at re- combination signal sequence (RSS) sites, then the breaks are repaired by the general double-stranded break repair mechanism. Depending on which J segment is chosen, there may be more than one left in the gene after the rearrangement. However, only the one closest to the V segment is used, and the others are spliced out of the primary transcript (by normal RNA splicing) in the process of connecting the C segment to the V and J for the final mRNA. Although this process generates great diversity, there is another mechanism that can generate further diversity under certain circumstances.
RAG1 and RAG2, lymphocyte-specific recombination activating genes, recognize the RSS sites and make the double-stranded cuts. Once the cuts are made, the excised portion joins its ends together to form a circular signal joint (SJ) which is then degraded. The coding portions have asymmetric cut ends that fold into hairpin formations and prevent their fusion. These hairpins are broken by Artemis, a nuclease recruited by DNA-dependent protein kinase (DNA-PK), which also brings together XRCC4, XLF, DNA ligase IV, and a DNA polymerase. The XRCC and XLF alight the DNA ends, recruits a terminal transferase that randomly adds nucleotides to the ends, then DNA polymerase λ or μ fills in the overhangs, and the ligase completes the join. Interestingly, this process adds even more variability to the immunoglobulin, since Artemis cuts the hairpin at random, and the terminal transferase also adds nucleotides at random.
Somatic hypermutation causes rearranged V segments to mutate at 105 times the rate of other DNA! This mechanism is carried out by Activation-Induced Cytidine Deaminase (AID), which converts cytidines to uracils, generating a G:U mismatch that is “corrected” by repair polymerases without strong error-correction. This hypermutation is initiated by the activation of a B cell by recognizing and binding to a ligand. As we will see in the next paragraphs, when that happens, the B cell initiates rapid proliferation and this is when the somatic hypermutation takes effect, so that many of the B cells will carry highly similar but subtly different antibodies than the initial B cell that recognized and was activated by the antigen. The idea is some of these subtle mutation may lead to antibodies with higher affinity for the antigen and therefore faster response the next time this particular pathogen tries to infect the organism.
The reason that this kind of DNA rearrangement is necessary is that antibody “design” is not a reactive system, but a proactive system. A common misconception is that the immune system encounters a pathogen and creates antibodies that t it. Unfortunately, there is no known mechanism by which a cell can “feel” the shape of something and create a protein that matches it. Instead, the immune system preemptively makes as many different antibodies (and TCRs) as possible, so that initially, most of the B and T cells in the body are actually genetically different. If an infection occurs, most of the B and T cells will bump into the pathogen and bounce right off, not recognizing it, but some of them will have the right antibody combination to bind to a part of the pathogenic invader. Although B cells can recognize surface antigens on their own, in most cases, a helper T cell is needed to activate the B cell (Figure \(\PageIndex{21}\)).

This process is initiated by a macrophage non-specifically ingesting a pathogen (1), breaking it apart, and presenting bits of it on its cell surface in partnership with MHC (major histocompatibility complex). A helper T-cell with a TCR that can recognize the antigen presented by the macrophage binds to it (2) and that leads to activation of the T cell. The activated helper T-cell binds to and activates a B cell (3) that has also bound to the antigen of interest, leading to massive B-cell proliferation (4), thus providing the body with many more copies of cells that have the right antibody to locate and fught the infection. This does two things: it provides lymphocyte reinforcements to specifically deal with a particular pathogen (but not other B cells, Figure \(\PageIndex{22}\)), and once the pathogen has been eliminated, there is a larger circulating pool of these cells to respond more quickly to any subsequent infection by this particular type of pathogen.

Finally, some of the B cells will differentiate into plasma cells (Figure \(\PageIndex{21}\)-5), that secrete antibodies into the blood- stream to provide a humoral response. Others become memory cells, which are like B cells in that the antibody is on its cell surface and not secreted, but they can be thought of as “pre-activated” and can respond more quickly than naive B cells to re-infection.
The big question that should have been lurking in the back of your mind through all this is, how do the antibodies and T-cell receptors tell what’s foreign and what’s part of one’s own body? We’ll get to that shortly. First, recall the activation of the helper T-cell. It occurs when the T-cell receptor recognizes an antigen from an ingested pathogen being presented by the MHC class II molecule on an antigen presenting cell (e.g. a macrophage).

Figure \(\PageIndex{23}\) shows the pathway from ingestion of the pathogen to the presentation of its molecular parts on an MHC molecule. A similar pathway also applies to presentation of antigens on MHC class I molecules in conjunction with cytotoxic (killer) T cells. The cytotoxic T cells work against compromised cells, whether they are infected by a virus or another pathogen (Figure \(\PageIndex{24}\)).

There are two major pathways to killing the infected cell. One is the activation of a “death receptor”, Fas, which induces a signal transduction cascade to activate caspases and apoptosis. The other pathway is the release of granzymes and perforins. The perforins drill into the membrane of the target cell and become pores that allow, among other things, granzymes to enter the cell, where they activate caspases by proteolysis, and again induce apoptosis. An important part of this is that the T cell receptor recognizes the antigen in combination with the MHC molecule that is presenting it. Furthermore, there are many variations of MHC molecules due there being 6 loci each with many known alleles.
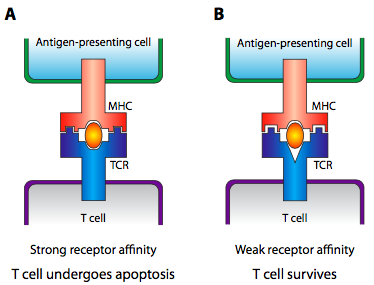
So, what does any of this have to do with self vs non-self recognition? Early in the development of the immune system, the MHC does not present bits of digested pathogens, it presents bits of the organism’s own cells that have gone through a proteasome or lysosome. At this early time, T cells behave somewhat differently, and if the T cell receptor binds strongly, the T cell commits apoptosis. This gets rid of TCR genes that strongly react to the organism’s own cells. If the T cells do not react at all, apoptosis is also invoked, because the TCR genes that cannot recognize the MHC will not be useful in an immune response. Only those T cells that very weakly bind to the self-presenting MHC survive (Figure \(\PageIndex{25}\)).