
12.9: Cell Motility
- Page ID
- 19242
There are a number of ways in which a cell can move from one point in space to another. In a liquid medium, that method may be some sort of swimming, utilizing ciliary or flagellar movement to propel the cell. On solid surfaces, those mechanisms clearly will not work efficiently, and the cell undergoes a crawling process. In this section, we begin with a discussion of ciliary/flagellar movement, and then consider the more complicated requirements of cellular crawling.
Cilia and flagella, which differ primarily in length rather than construction, are microtubule-based organelles that move with a back-and-forth motion. This translates to “rowing” by the relatively short cilia, but in the longer flagella, the flexibility of the structure causes the back-and-forth motion to be propagated as a wave, so the flagellar movement is more undulating or whiplike (consider what happens as you waggle a garden hose quickly from side to side compared to a short piece of the same hose). The core of either structure is called the axoneme, which is composed of 9 microtubule doublets connected to each other by ciliary dynein motor proteins, and surrounding a central core of two separate microtubules.
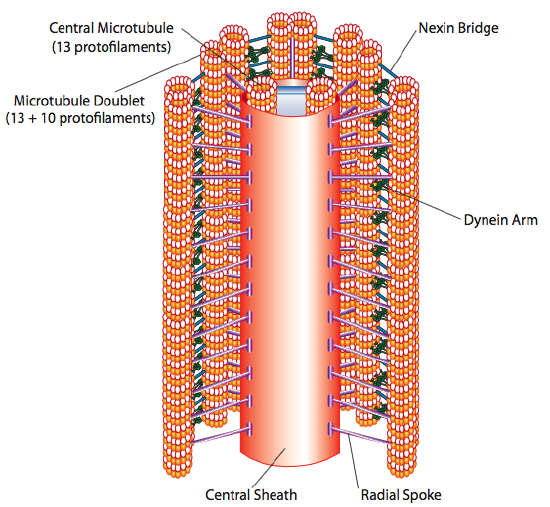
This is known as the “9+2” formation, although the nine doublets are not the same as the two central microtubules. The A tubule is a full 13-protofilaments, but the B tubule fused to it contains only 10 protofilaments. Each of the central microtubules is a full 13 protofilaments. The 9+2 axoneme extends the length of the cilium or flagellum from the tip until it reaches the base, and connects to the cell body through a basal body, which is composed of 9 microtubule triplets arrange in a short barrel, much like the centrioles from which they are derived.
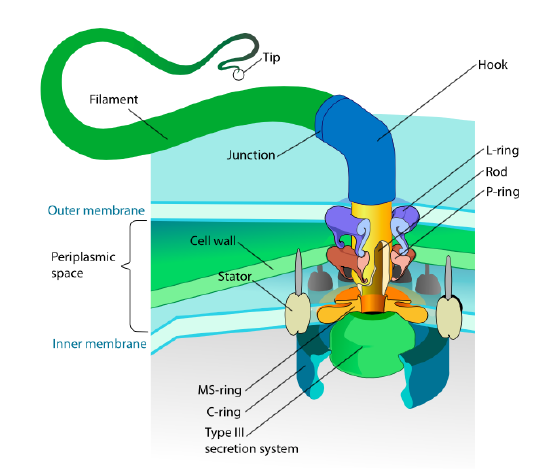
This section refers only to eukaryotes. Some prokaryotes also have motile appendages called flagella, but they are completely different in both structure and mechanism. The flagella themselves are long helical polymers of the protein flagellin, and the base of the flagellin fibers is connected to a rotational motor protein, not a translational motor. This motor (Figure \(\PageIndex{18}\)) utilizes ion (H+ or Na+ depending on species) down an electrochemical gradient to provide the energy to rotate as many as 100000 revolutions per minute. It is thought that the rotation is driven by conformational changes in the stator ring, nestled in the cell membrane.
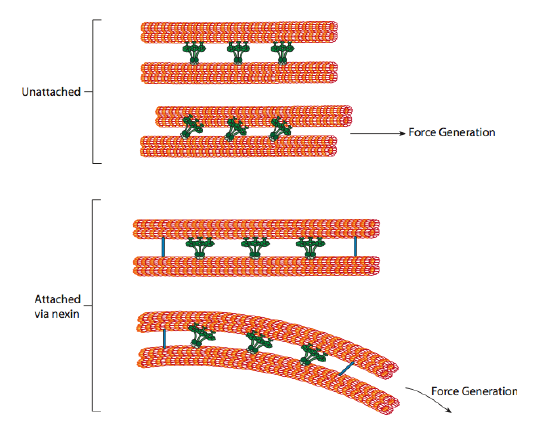
The ciliary dyneins provide the motor capability, but there are two other linkage proteins in the axoneme as well. There are nexins that join the A-tubule of one doublet to the B-tubule of its adjacent doublet, thus connecting the outer ring. And, there are radial spokes that extend from the A tubule of each doublet to the central pair of microtubules at the core of the axoneme. Neither of these has any motor activity. However, they are crucial to the movement of cilia and flagella because they help to transform a sliding motion into a bending motion. When ciliary dynein (very similar to cytoplasmic dyneins but has three heads instead of two) is engaged, it binds an A microtubule on one side, a B microtubule from the adjacent doublet, and moves one relative to the other. A line of these dyneins moving in concert would thus slide one doublet relative to the other, if (and it’s a big “if”) the two doublets had complete freedom of movement. However, since the doublets are interconnected by the nexin proteins, what happens as one doublet attempts to slide is that it bends the connected structure instead (Figure \(\PageIndex{17}\)). This bend accounts for the rowing motion of the cilia, which are relatively short, as well as the whipping motion of the long flagella, which propagate the bending motion down the axoneme.
Although we think of ciliary and flagellar movement as methods for the propulsion of a cell, such as the flagellar swimming of sperm towards an egg, there are also a number of important places in which the cell is stationary, and the cilia are used to move fluid past the cell. In fact, there are cells with cilia in most major organs of the body. Several ciliary dyskinesias have been reported, of which the most prominent, primary ciliary dyskinesia (PCD), which includes Kartagener syndrome (KS), is due to mutation of the DNAI1 gene, which encodes a subunit (intermediate chain 1) of axonemal (ciliary) dynein. PCD is characterized by respiratory distress due to recurrent infection, and the diagnosis of KS is made if there is also situs inversus, a condition in which the normal left-right asymmetry of the body (e.g. stomach on left, liver on right) is reversed. The first symptom is due to inactivity of the numerous cilia of epithelial cells in the lungs. Their normal function is to keep mucus in the respiratory track constantly in motion. Normally the mucus helps to keep the lungs moist to facilitate function, but if the mucus becomes stationary, it becomes a breeding ground for bacteria, as well as becoming an irritant and obstacle to proper gas exchange.
Situs inversus is an interesting malformation because it arises in embryonic development, and affects only 50% of PCD patients because the impaired ciliary function causes randomization of left-right asymmetry, not reversal. In very simple terms, during early embryonic development, left-right asymmetry is due in part to the movement of molecular signals in a leftward ow through the embryonic node. This flow is caused by the coordinated beating of cilia, so when they do not work, the flow is disrupted and randomization occurs.
Other symptoms of PCD patients also point out the work of cilia and flagella in the body. Male infertility is common due to immotile sperm. Female infertility, though less common, can also occur, due to dysfunction of the cilia of the oviduct and fallopian tube that normally move the egg along from ovary to uterus. Interestingly there is also a low association of hydrocephalus internus (overfilling of the ventricles of the brain with cerebrospinal fluid, causing their enlargement which compresses the brain tissue around them) with PCD. This is likely due to dysfunction of cilia in the ependymal cells lining the ventricles, and which help circulate the CSF, but are apparently not completely necessary. Since CSF bulk flow is thought to be driven primarily by the systole/ diastole change in blood pressure in the brain, some hypothesize that the cilia may be involved primarily in ow through some of the tighter channels in the brain.
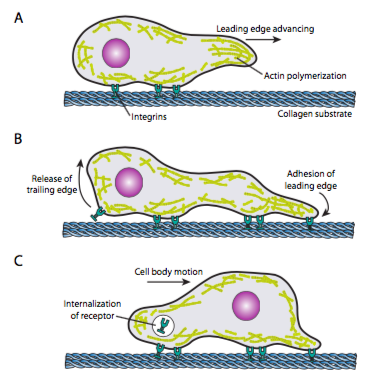
Cell crawling (Figure \(\PageIndex{19}\)) requires the coordinated rearrangement of the leading edge microfilament network, extending (by both polymerization and sliding filaments) and then forming adhesions at the new forward-most point. This can take the form of filopodia or lamellipodia,and often both simultaneously. Filopodia are long and very thin projections with core bundles of parallel microfilaments and high concentrations of cell surface receptors. Their purpose is primarily to sense the environment. Lamellipodia often extend between two lopodia and is more of a broad ruffle than a finger. Internally the actin forms more into meshes than bundles, and the broader edge allows for more adhesions to be made to the substrate. The microfilament network then rearranges again, this time opening a space in the cytoplasm that acts as a channel for the movement of the microtubules towards the front of the cell. This puts the transport network in place to help move intracellular bulk material forward. As this occurs, the old adhesions on the tail end of the cell are released. This release can happen through two primary mechanisms: endocytosis of the receptor or deactivation of the receptor by signaling/conformational change. Of course, this oversimplification belies the complexities in coordinating and controlling all of these actions to accomplish directed movement of a cell.
One model of microfilament force generation, the Elastic Brownian Ratchet Model (Mogilner and Oster, 1996), proposes that due to Brownian motion of the cell membrane resulting from continuous minute thermal fluctuation, the actin filaments that push out towards the edges of the membrane are flexed to varying degrees. If the flex is large enough, a new actin monomer can fit in between the membrane and the tip of the filament, and when the now longer filament flexes back, it can exert a greater push on the membrane. Obviously a single filament does not generate much force, but the coordinated extension of many filaments can push the membrane forward.
Once a cell receives a signal to move, the initial cytoskeletal response is to polymerize actin, building more microfilaments to incorporate into the leading edge. Depending on the signal (attractive or repulsive), the polymerization may occur on the same or opposite side of the cell from the point of signal-receptor activation. Significantly, the polymerization of new f-actin alone can generate sufficient force to move the membrane forward, even without involvement of myosin motors! Models of force generation are being debated, but generally start with the incorporation of new g-actin into a filament at its tip; that is, at the filament-membrane interface. Even if that might technically be enough, in a live cell, myosins are involved, and help to push and arrange filaments directionally in order to set up the new leading edge. In addition, some filaments and networks must be quickly severed, and new connections made, both between filaments and between filaments and other proteins such as adhesion molecules or microtubules.
How is the polymerization and actin rearrangement controlled? The receptors that signal cell locomotion may initiate somewhat different pathways, but many share some commonalities in activating one or more members of the Ras-family of small GTPases. These signaling molecules, such as Rac, Rho, and cdc42 can be activated by receptor tyrosine kinases (see RTK-Ras activation pathways, Chap. 14). Each of these has a slightly different role in cell motility: cdc42 activation leads to filopodia formation, Rac activates a pathway that includes Arp2/3 and cofilin to lamellipodia formation, and Rho activates myosin II to control focal adhesion and stress fiber formation. A different type of receptor cascade, the G-protein signaling cascade (also Chapter 14), can lead to activation of PLC and subsequent cleavage of PIP2 and increase in cytosolic Ca2+. These changes, as noted earlier, can also activate myosin II, as well as the remodeling enzymes gelsolin, cofilin, and profilin. This breaks down existing actin structures to make the cell more fluid, while also contributing more g-actin to form the new leading edge cytoskeleton.
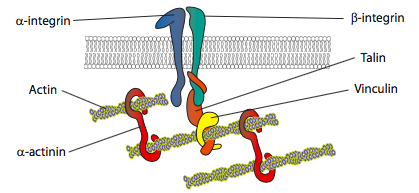
In vitro experiments show that as the membrane pushes forward, new adhesive contacts are made through adhesion molecules or receptors that bind the substrate (often cell culture slides or dishes are coated with collagen, filaminin, or other extracellular matrix proteins). The contacts then recruit cytoskeletal elements for greater stability to form a focal adhesion (Figure \(\PageIndex{20}\)). However, the formation of focal adhesions appears to be an artifact of cell culture, and it is unclear if the types of adhesions that form in vivo recruit the same types of cytoskeletal components.
The third step to cell locomotion is the bulk movement of the cellular contents forward. The mechanisms for this phase are unclear, but there is some evidence that using linkages between the actin cytoskeleton at the leading edge and forward parts of the microtubule cytoskeleton, the microtubules are rearranged to form an efficient transport path for bulk movement. Another aspect to this may be a “corralling” effect by the actin networks, which directionally open up space towards the leading edge. The microtubules then enter that space more easily than working through a tight actin mesh, forcing flow in the proper direction.
Much of the work on microtubule-actin interactions in cell motility has been done through research on the neuronal growth cone, which is sometimes referred to as a cell on a leash, because it acts almost independently like a crawling cell, searching for the proper pathway to lead its axon from the cell body to its proper synaptic connection (A.W. Schaefer et al, Dev. Cell 15: 146-62, 2008).
Finally, the cell must undo its old adhesions on the trailing edge. This can happen in a number of different ways. In vitro, crawling cells have been observed to rip themselves off of the substrate, leaving behind tiny bits of membrane and associated adhesion proteins in the process. The force generated is presumed to come from actin-myosin stress fibers leading from the more forward focal adhesions. However, there are less destructive mechanisms available to the cells. In some cases, the adhesivity of the cellular receptor for the extracellular substrate can be regulated internally, perhaps by phosphorylation or dephosphorylation of a receptor. Another possibility is endocytosis of the receptor, taking it off the cell surface. It could simply recycle up to the leading edge where it is needed (i.e. transcytosis), or if it is no longer needed or damaged, it may be broken down in a lysosome.