
11.7: Receptor-mediated Endocytosis
- Page ID
- 19209
Just as there is vesicular traffic towards the plasma membrane, either for secretion or for incorporation of membrane lipids or proteins, there can also be vesicular traffic from the plasma membrane. Endocytosis is the process by which a coat protein (usually clathrin) on the cytoplasmic side of the plasma membrane, begins to polymerize a coat that draws the membrane with it into a vesicle. However, instead of capturing a bit of ER or Golgi lumen with it, the vesicle contains a little material from outside of the cell. Sometimes endocytosis is initiated internally, perhaps to remove a particular protein from the cell surface (for an example, see trailing edge dynamics in cell motility in the next chapter), but often, the endocytosis is the result of a ligand binding to an extracellular receptor molecule, leading to its activation and subsequent nucleation of a clathrin assembly and vesicle formation.
There are many types of ligands: a nutrient molecule (usually on a carrier protein, as in the examples below) or even an attacking virus which has co-opted the endocytic mechanism to facilitate entry into the cell. The example depicted here is a classic example: endocytosis of cholesterol (via low-density lipoprotein). This illustrates one potential pathway that the receptors and their cargo may take. In the case of cholesterol, the carrier protein is broken down fully, although in the case of transferrin, a serum protein that carries iron in the blood, the carrier protein is just recycled after releasing its transferrin cargo. It is packaged into an exocytic vesicle headed back to the cell surface.
Serum cholesterol is usually esterified and bound by LDL (low density lipoprotein), which then floats about in the bloodstream until it meets up with an LDL receptor on the surface of a cell. When the LDL binds to its receptor, the receptor is activated, and a clathrin-coated vesicle forms around the LDL/receptor complex. LDL receptors tend to aggregate in what are known as clathrin-coated pits — crater-like partial vesicles that already have a small number of polymerized clathrin molecules. The vesicle forms exactly as described previously for Golgi-derived clathrin vesicles: the clathrin self- assembles into a spherical vesicle, and dynamin pinches the vesicle off the cell membrane. This vesicle then fuses with an early endosome, which carries proton pumps in its membrane, causing the environment inside the vesicle to acidify (~pH 6). This acidification can cause conformational shifts in proteins that could, for example, lead to a receptor releasing its ligand, as is the case here with LDL and LDL receptor. The early endosome also functions as a sorting station: the receptor is re-vesicularized and transported back to the plasma membrane. Meanwhile, the LDL is packaged into a different vesicle and heads off for further processing.
The endosomal proton pumps are ATP-driven, Mg2+-dependent V-type pump (as opposed to the F-type pump in the mitochondrial inner membrane). Structurally, the two are similar though, and ATP hydrolysis drives the rotary unit, which then powers the movement of protons across the membrane from cytoplasm into endosome.
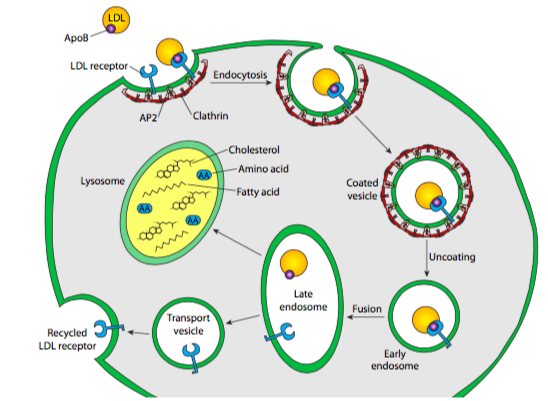
The endosomal vesicle with the LDL in it next fuses with another acidic, membrane-bound compartment. The lysosome, at pH ~5.0, is even more acidic than the endosome, and it also contains a large complement of acid hydrolases — hydrolytic enzymes ranging across substrates (including proteases, lipases, glycosidases, nucleases) that operate optimally in acidic conditions, and minimally in the neutral or slightly basic conditions in the cytoplasm. In part, this is a safety mechanism — leakage of digestive enzymes from the lysosome will not result in wholesale digestion of the cell because the enzymes have little or no activity in the cytoplasm. The lysosomal membrane, in addition to having proton pumps to acidify the internal environment, also incorporates many transporter proteins to aid in moving the digestion products of the acid hydrolases out of the lysosome so that the cell can make use of the amino acids, sugars, nucleotides, and lipids that result. Back to our example, that means that the cholesterol esters are broken apart into individual cholesterol molecules, and the lipoprotein is broken down into lipids and amino acids. Interestingly, these transporter proteins are not digested by the lysosomal proteases because they are very heavily glycosylated, which shields potential proteolytic sites from the proteases.
Lysosomal enzymes are specifically tagged by a mannose-6-phosphate that is added in the cis Golgi. This is a two-step process in which N-acetylglucosamine phosphotransferase adds a phospho-GlcNAc to a mannose residue, connecting via the phosphate group, then a phosphodiesterase removes the GlcNAc, leaving the mannose-6-P. This specifically targets lysosomal enzymes because they all have specific protein recognition sequences that the phosphotransferase binds to before transferring the P-GlcNAc. Although the lysosomal enzymes are tagged in the cis Golgi, they do not sort until the trans Golgi, when mannose-6-P receptors bind to the lysosomal enzymes and form lysosomal vesicles that will bud off and travel to late endosomes and lysosomes to deliver their acid hydrolase payload. Again, the pH change is important: in the somewhat acidic (pH 6.5) environment of the trans Golgi, the receptor binds the mannose-6-P-tagged enzymes, but in the more acidic lysosome, the acid hydrolases are released to do their work.
When one or more acid hydrolases do not function properly or do not make it into the lysosome due to improper sorting, the result is incomplete digestion of the lysosomal contents. This in turn leads to the formation of large inclusions of partially digested material inside the lysosomes. This accumulation of material can be cytotoxic, and genetic disorders that affect the expression or sorting of lysosomal hydrolases are collectively referred to as lysosomal storage diseases. These fall into several categories depending on the types of molecules accumulated.
A common and easily treatable disease of glycosaminoglycan accumulation is Hurler’s disease, which can be effectively treated and non-neurological effects even reversed by enzyme replacement therapy. Hurler’s others in its class affect a wide variety of tissues because glycosaminoglycans are ubiquitous. On the other hand, because the brain is enriched in gangliosides, lysosomal storage diseases like Gaucher’s disease show defects primarily in the CNS. Many lysosomal storage diseases have similar presentation: developmental abnormalities, especially stunted bone growth, lack of fine facial features, and neuromuscular weakness.
Since it depends greatly on the contents of the endosome(s) that fused with it, the size and contents of lysosomes can vary greatly. In fact, the lysosome may also degrade internal cellular components through the process of autophagy. Usually, this is initiated under starvation conditions which lead to inhibition of mTor, and subsequent expression of autophagic genes. These then interact with mitochondria and other cellular components, and promote the formation of a double-membraned autophagosome around them. The origin of the membranes is unclear, although the ER is suspected. Finally, the autophagosome fuses with a lysosome, and the acid hydrolases break down the cell parts for energy. A variation on this called microautophagy can also occur, in which the lysosome itself invaginates a bit of cytoplasmic material and internalizes an intralysosomal vesicle that is then broken down.
The most severe, I-cell disease (mucolipidosis type II) occurs when nearly all lysosomal enzymes are missing in the fibroblasts of the affected individual. There is severe developmental delay and early growth failure, neuromuscular problems, and malformations in early skeletal development. The severity of this disorder is due to the almost complete lack of lysosomal enzymes, which is caused by a deficiency of GlcNAc phosphotransferase. Without it, no enzymes are tagged for sorting to the lysosome.
Other relatively common disorders include Tay-Sachs and Niemann-Pick diseases. Tay-Sachs is caused by an accumulation of gangliosides in the brain and is usually fatal by 5 years of age. Niemann-Pick, on the other hand, may manifest as Type A with an even shorter life expectancy, or as Type B, in which symptoms are relatively minor. The major difference is that Type A patients have very little (<5%) of their sphingomyelinase activity, while Type B patients have only slightly less than normal (~90%) activity.
Finally, it should be noted that the large vacuoles of plant cells are in fact specialized lysosomes. Recall that vacuoles help to maintain the turgor, or outward water pressure on the cell walls that lead to a rigid plant part rather than a limp, wilted one. One of the ways in which this occurs is that the acid hydrolases inside the vacuole alter the osmotic pressure inside the vacuole to regulate the movement of water either in or out.
Another example of receptor-mediated endocytosis is the import of iron into a mammalian cell. As with serum cholesterol, iron is not generally imported into the cell by itself. Instead, it is bound to apotransferrin, a serum protein that binds two Fe3+ ions. Once it has bound the iron ions, the apotransferrin is now referred to as transferrin, and it can be recognized and bound by transferrin receptors (TfR) located on the extracellular surface of cell membranes. This initiates receptor-mediated endocytosis just as described above. However, in this case, the lysosome is not involved. As the transferrin and transferrin receptor reach the early endosome, they do not dissociate, but rather the Fe2+ releases from the transferrin, and then exits the endosome via DMT1, a divalent metal transport protein to be used in heme groups or other complexes. This leaves the apotransferrin-TfR complex, which is recycled back to the cell membrane via vesicle. Once the vesicle fuses with the extracellular space, the acidity of the endosome is dissipated and the apotransferrin no longer binds to TfR. Apotransferrin can thus go back to its duty of finding iron ions and bringing them back to the cell.