
7.6: DNA Repair
- Page ID
- 16133

Strictly defined, the simplest repair mechanism does not use an enzyme. Dealkylation, or removal of alkyl groups (like —CH3 or —C2H5) involves only the transfer of an alkyl group from an O6-methylguanine or O6-ethylguanine onto O6-alkylguanyl-DNA alkyltransferase. Despite the name, the alkyltransferase is not really an enzyme, since it is permanently altered and inactivated by the reaction and therefore does not fit the definition of a catalyst. Note that this does not remediate alkylation at N7 or other sites, just the O6-linked ones.
The next simplest repair mechanism is the uncoupling of pyrimidine cyclobutyl dimers. This can be accomplished through the activity of DNA photolyases, also known as photoreactivating enzymes. These are named not just because the formation of the pyrimidine cyclobutyl dimers is usually due to UV light exposure, but because the repair enzymes themselves require exposure to light (300-500 nm, near UV to visible blue) to catalyze the dimer-breaking reaction.
More specifically, DNA photolyases (a ~60kD protein), are non-covalently associated with a chromophore (N5,N10-methylenyltetrahydrofolate or 5-deazaflavin) and an FADH—. The photolyase binds to the pyrimidine cyclobutyl dimer of either single-stranded or double-stranded DNA in a light-independent and sequence-independent manner. However it does not catalyze any change in the bond until light is absorbed by the chromophore, which then transfers the energy to FADH—, causing it to eject and electron to the dimer, thus breaking it apart.
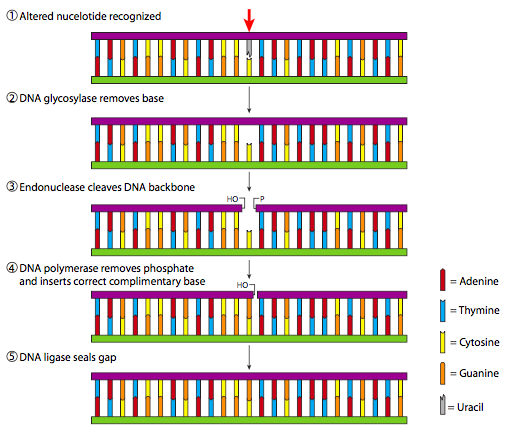
While dealkylation and dimer lysis are relatively simple processes that make only a subtle change to the DNA, excision repair mechanisms are more complicated and require multiple enzymatic steps to complete. When a small (not sterically bulky) lesion is limited to a single base, whether missing from depurination or incorrectly formed due to deamination or misincorporation, the process known as base excision repair (BER) is engaged. As illustrated in Figure \(\PageIndex{20}\), if a non-conventional base is recognized, it is then removed by an appropriate DNA glycosylase. At present (Genbank search, July 2009), there are at least 8 specific genes encoding human DNA glycosylases, although three encode glycosylases that recognize uracil in various situations. Once the base has been removed by the glycosylase, an endonuclease is enlisted to break the phosphodiester bonds than hold the now-empty phosphodeoxyribose. The resulting gap in the DNA is filled in by a DNA polymerase and finally the strand is reconnected by DNA ligase.
In the case of bulky lesions that significantly alter the physical presentation of the DNA to the polymerases and other enzymes that process DNA, a different type of repair process is involved. Nucleotide excision repair (NER), perhaps better named polynucleotide excision repair, involves the removal of the lesion as well as some of the nucleotides in the immediate vicinity. There are two major initiators of NER: either a non-transcriptionally active portion of the DNA is scanned by XPC (Figure \(\PageIndex{21}\)A), which recognizes a bulky lesion and recruits the repair complex, or as a gene is being transcribed, RNA polymerase runs into a lesion, and then recruits the repair complex via CSA and CSB (Figure \(\PageIndex{21}\)F and G). If the detection is through XPC, one of the early repair factors recruited to the site is Transcription Factor IIH/XPB/XPD, which is a DNA helicase (Figure \(\PageIndex{21}\)B). This type of global genome detection is inefficient and relatively slow, but provides a basal level of error-checking for all DNA. In the case of DNA being transcribed, the RNA polymerase complex already includes TFIIH, of which XPB and XPD are a part. This transcriptionally-directed detection is more efficient and targets those parts of the DNA in greatest use in a given cell. In the next step (Figure \(\PageIndex{21}\)C), XPG, associated with BRCA1/2, and XPF, associated with ERCC1, excise a portion of the affected strand, including but not limited to the lesion itself. DNA polymerase δ or ε can then add onto the free 3’OH to ll in the gap based on the complementary strand sequence (Figure \(\PageIndex{21}\)D). Finally, the repair is connected on its 3’ end to the rest of the strand by DNA ligase (Figure \(\PageIndex{21}\)E).
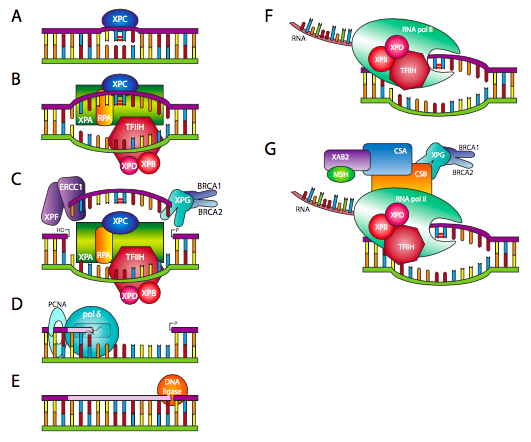
The “XP” in XPC, XPB, XPD, and the others in Figure \(\PageIndex{21}\) refers to xeroderma pigmentosa, another autosomal recessive disease, of which the primary characteristic is the formation of skin carcinomas at a young age. Because NER is a major form of pyrimidine dimer repair (in addition to photolyases), its disruption by mutations to one or more of the XP genes leads to extreme sensitivity to UV-induced lesions. Affected individuals must minimize exposure to the sun. The name of the disease comes from the characteristic pigmented lesions (keratoses) that often form on the skin when exposed to sun.
CSA and CSB are named for Cockayne syndrome, an autosomal recessive aging disorder. Mutations in either gene can cause the disorder, which is characterized by premature aging, stunted growth, photosensitivity, and developmental defects of the nervous system. Presumably, knocking out the DNA repair capability of CSA or CSB leads to fast accumulation of damage, inability to transcribe needed genes, and eventually cell death.
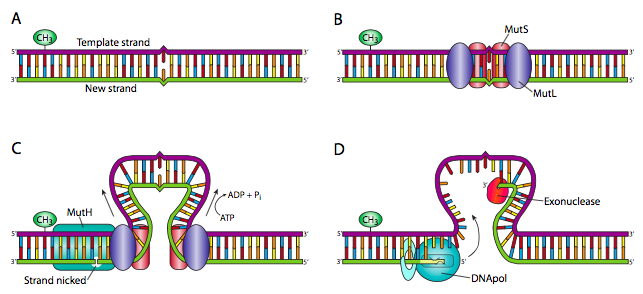
A sort of variation on NER is the mismatch repair (MMR) system. This is best understood in prokaryotes: in E. coli, MutS is a small protein that forms homodimers at mismatch sites. The MutS dimers recruit two MutL proteins, each of which interacts with one of the MutS units. Each MutS/MutL complex pushes DNA through inwardly, forming a loop with the mismatch in the center of the loop. This continues until one of the MutS/MutL complexes encounters a hemimethylated GATC sequence. This causes recruitment of MutH, a highly specialized endonuclease that makes a single-stranded nick in the backbone of the non-methylated strand. This provides an opening for the 3’-5’ exonuclease I or the 5’-3’ exonuclease VII (or RecJ) to degrade the strand from the nick to the point of mismatch. This is then, as you may have guessed, filled in by DNA polymerase and the backbone connected by ligase. In eukaryotes, multiple homologues to the MutS and MutL proteins have been discovered and the process is similar, but not clearly understood yet, as no homologue to MutH has yet been discovered.
Recall that in E. coli, Dam methyltransferases eventually methylate the DNA as a method of protecting its genome, but newly synthesized DNA is not methylated. Thus, the assumption is that the methylated strand contains the original and correct base, while the mismatch is due to misincorporation in the newer strand.
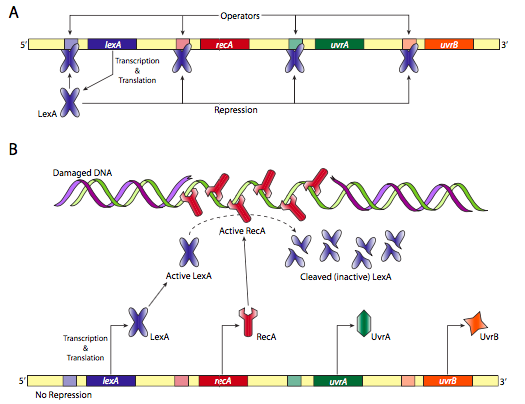
Another prokaryotic DNA repair system is the SOS response. As depicted in Figure \(\PageIndex{23}\) below, if there is no damage, RecA is inactive, so LexA protein can repress the production of more SOS repair proteins. However, if there is damage, RecA proteins bind to the single stranded DNA and are activated. They in turn cleave the LexA repressor allowing production from a number of DNA repair genes.
So far, the repairs have been based on the assumption that a lesion affects only one strand, and the other strand can provide a reliable template for effecting repairs without the loss of information. Unfortunately, that is not always the case, and some lesions and repair processes necessarily lead to sequence loss. When a double-strand break occurs, perhaps as the product of ionizing radiation, the most common repair mechanism is known as non-homologous end joining (NHEJ). The double-stranded ends are first recognized by Ku, a heterodimeric circular protein that binds the DNA ends. Ku then recruits the kinase DNA-PKCS. The DNA-PKCS acts as a bridge to bring the two ends together, and a DNA ligase can then join the ends together. If the strands were broken in different places, resulting in complementary single-stranded overhangs at each end (like those generated by some restriction endonucleases) then the repair is often perfect, since the complementary sequences align the two ends correctly in their original positions. However, if the strand ends have already been acted upon by nucleases and are no longer complementary, then the rejoining of the ends will likely lead to loss of information. In some parts of the DNA, this would have little effect, but if it happened within a gene, the mutated gene product could have abnormal or compromised function.